Tek Umblikal Arter Kadınlar Kulübü
GÖBEK KORDONUNDA TEK ARTER TEK VEN
Umblikal kordon içindeki 2 arterden sağ veya soldakinin olmamasıdır. Fetus göbek kordonun içinde 1 venve 2 arter bulunur. Bu arterlerin biri mesanenin sağ, diğeri ise sol yanından gelerek kordon içine girip vena ile birlikte sarmal şekilde devam ederler. Tek umblikal arter anomalisinde ise 1 ven ve 1 arter vardır. Bu 2 arterde bebekte bulunan oksijenlenmesi gereken pis kan plasentaya (çocuğun eşi) gider ve orada oksijenlenir . Temiz kan ven ile bebeğe geri döner . Ven bebekte karaciğere ve kalbe , kalptende beyine ilk temiz kanı getirir. Normal fetuslarda plasentaya olan kan akımı her iki arterden eşit miktarda olur. Tek arter olduğunda ise mevcut arter, olmayan diğer arterin görevini de üstlenir.
Yenidoğan bebeklerde görülen umblikal kordona ait en yaygın anomalidir. Yapılan araştırmalarda tek umblikal arter’in görülme sıklığı; % 0.08 ile %1.9 gibi değişik değerlerde bildirilmiştir.
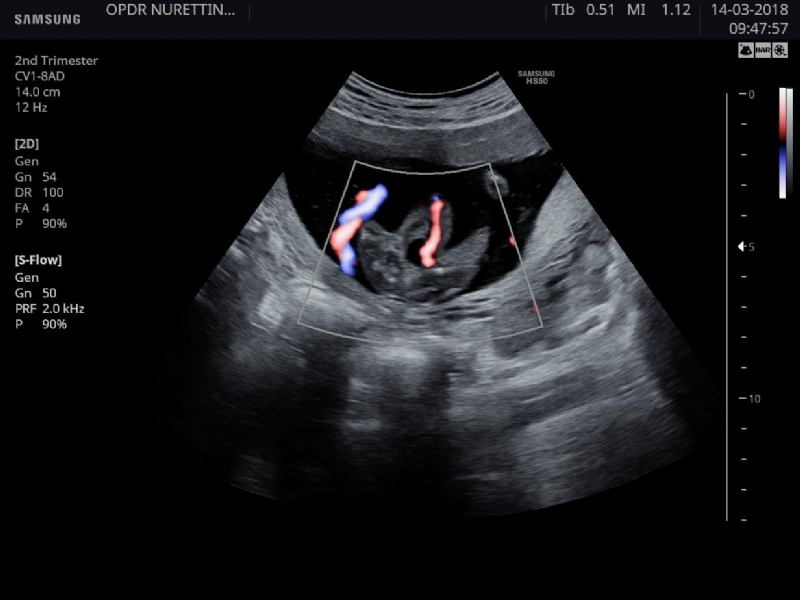
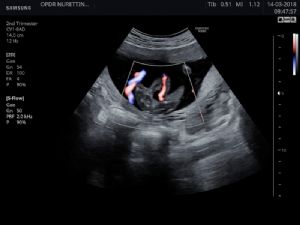
Ultrasonografik değerlendirmede umblikal kordon kesitinde 2 damarın görülmesi gerekir. Normalde geniş çaplı 1 vena ve daha dar çaplı 2 adet arter görülür. Tek umblikal arterde ise arterin çapı genişlemiştir. Normal kordon yapısındaki arterlerin her birinin çapı diğerine eşittir ve arter çapları vena çapının yarısından daha azdır. Tanıya yardımcı olan diğer bir bulgu ise fetal pelvisin transvers kesitinde renkli dopplerle mesanenin tek tarafından umblikal arter gelişinin görülmesidir. Oysa normal umblikal arter yapısı olduğunda mesanenin iki tarafinda ayrı ayrı umblikal arterlerin görüntülenmesi mümkündür. Sol umblikal arter yokluğunun %70, sağ umblikal arter yokluğunun ise %30 sıklıkla görüldüğü bildirilmiştir.
Tek umblikal arter saptanan olgularda ilave anomalileri varlığı da arştırılmalıdır. Çünkü bu olgularda sıklıkla ilave anomalilere rastlanmaktadır. Bu anomalilerin en sık görülenleri; plasental yapı anomalileri, fetal santral sinir sistemi (SSS) anomalileri, yüz anomalileri, ekstremite anomalileri, kalp anomalileri, böbrek anomalileri, Trizomi-13, Trizomi 18 ve kromozom anomalileridir. Tek umblikal arter varlığında ilave anomali olsun veya olmasın olguların yaklaşık %15-%20 sinde fetal büyüme-gelişme geriliği görülür.
Tek umblikal arter görüldüğünde ilave yapısal anomalide varsa bu olguların yaklaşık %50’lik bölümünde özellikle Trizomi 13 ve Trizomi 18 başta olmak üzere kromozomal anomaliler görülebilir. Bu nedenle bu olgularda genetik değerlendirme gerekir. Bu amaçla amniosentez (AS) ile kromozomal analiz yapılması önerilir.
Bunların yaklaşık %15- %20’lik bölümünde fetal gelişme geriliği ortaya çıkabilir. Bu nedenle bu gebeliklerin sık aralıklarla kontrol edilmesi ve 3. trimesterin başından itibaren fetal takipte umblikal arter Doppler değerlendirilmesinin de ilave edilmesi gerekir.
Doppler Testi
Renkli Doppler ultrasonografi
 Doppler, genel anlamda ultrason altında incelenen damar yatağının kan akımını ve normaldışı bir dirençle karşı karşıya olup olmadığını belirleyen bir testtir.
Doppler, genel anlamda ultrason altında incelenen damar yatağının kan akımını ve normaldışı bir dirençle karşı karşıya olup olmadığını belirleyen bir testtir.
Kalbin atım ve dolum fazında incelenen damar yatağındaki basınçlar karşılaştırarak direnç belirlenir.
Damarın inceleme esnasında ekranda oluşturduğu dalga görüntüsü de damar yatağının önündeki direnç hakkında bilgi verir.
Obstetrik (gebelikle ilgili) uygulamalarda anneden bebeğe kan götüren uteroplasental üniteyi değerlendirebileceği gibi, bebeğin kordonun kan akımı konusunda bilgi verir.
Video: GEBELİKTE RENKLİ DOPPLER İLE ANNE ADAYI VE BEBEKTE HANGİ SORUNLAR ANLAŞILIR VE TEDAVİSİ VAR MIDIR?
Dr. Kağan Kocatepe Youtube Kanalı >>Yüksek rizikolu gebeliklerin takibinin önemli bir parçasıdır.
Hangi durumlarda uygulanır?
Umbilikal arterdeki (göbek kordonu) akım ölçümlerinin patolojik çıkması ya da anormal dalga şekillerinin gözlenmesi bebekte ölçüm esnasında fetal distres varlığına işaret edebileceği gibi, bebekte gebeliğin ilerleyen dönemlerinde gelişme geriliği (IUGG) ya da fetal distres gelişme riskinin arttığına işaret edebilir. Ayrıca incelemedeki çeşitli patolojiler bebekteki bir kromozom anomalisi varlığı konusunda şüphe uyandırabilir.
Video: UMBİLİKAL ARTER RENKLİ DOPPLERİ NASIL VE NEDEN YAPILIR? KİLO ALAMAYAN BEBEKLERDE NEYİ GÖSTERİR?
Dr. Kağan Kocatepe Youtube Kanalı >>Uterin arter dopplerinde direnç artışı ya da dalga görünümünde anormallik (çentiklenme) ise anne adayında gebeliğin ileri dönemlerinde preeklampsi gelişeceğini gösterebilir.
Video: RENKLİ DOPPLER NASIL VE NEDEN YAPILIR, UTERİN ARTERDE ÇENTİK (NOTCH) VE DİRENÇ ARTIŞININ RİSKİ NEDİR?
Dr. Kağan Kocatepe Youtube Kanalı >>Bu haliyle umbilikal ve uterin arter doppler incelemeleri en sık preeklampsili ve kronik hipertansiyonlu anne adaylarının izlenmesinde kullanılır. Ayrıca kontrolsüz diyabetli anne adaylarında, polihidramnioslu (bebeğin sıvısının artması) ya da oligohidramnioslu (bebeğin sıvısının azalması) anne adaylarında, fetuslardan birinde gelişim kusuru olan çoğul gebeliklerde yaklaşım biçimini oluşturmakta yardımcı bir yöntem olarak kullanılabilir.
Bunun dışında 23.-24. gebelik haftalarında yapılan ayrıntılı ultrasonun bir parçası olarak da, Doppler uygulayan ekoller ve doktorlar vardır. Doppler incelemesinin rutin olarak tüm anne adaylarına uygulanmasının gerekli olup olmadığı henüz açıklık kazanmış bir durum değildir.
Uygulanması
Anne adayına yapılan rutin ultrason incelemesinden sonra önce sağlı sollu uterin arterlerde (rahim atardamarlarında), sonra da umbilikal (kordon) arterde Doppler incelemesi yapılır. Her iki uterin arterin akım değerleri, ikisi arasındaki matematiksel fark ve görünüm şekilleri değerlendirilir. Umbilikal arterde de yine akım değerlendirilir ve dalga şekli incelenir.
Video: Detaylı ultrasonografi ve renkli Doppler incelemesi nasıl yapılır?
Yorumlanması
Doppler tanı koydurucu değil yönlendirici bir testtir. Patolojik akım değerleri ya da anormal dalga görünümleri elde edildiğinde bebeğin diğer iyilik hali testleriyle yakından değerlendirilmesi konusunda hassas olunması gerektiğini gösterir.
Bunun tek istisnası umbilikal arterde "ters akım" denen bir durumun ortaya çıkmasıdır. Umbilikal arterde bu anormal dalga şekli gözlendiğinde bebeğin karında ölme riski oldukça yüksektir ve acil doğum gerekebilir.
Video: 20. gebelik haftasında uterin renkli Doppler nasıl yapılır? - Direnç artışı ve notch (çentik) nedir?
Dr. Kağan Kocatepe Youtube Kanalı >>
Tanıtım Videosu

Tanıtım Kataloğu

Sanal Tur
Hastalık Tanımı
Glukokortikoid eksikliği, hiperandrojenizm, hipertansiyon ve kadınlarda virilizasyon ile karakterize nadir görülen bir konjenital adrenal hiperplazi (KAH) formudur.
ORPHA: 90795
Eş Anlamlısı(ları):
11-beta-hidroksilaz eksikliğine bağlı KAH
CYP11B1 eksikliği
Prevalans: 1-9/1,000,000
Kalıtım: Otozomal resesif
Başlangıç yaşı: Bebeklik, Yenidoğan
ICD-10: E25.0
OMIM: 202010
UMLS: C0268292
MeSH: C535978
GARD: 5658
MedDRA: 10000002
Özet
Epidemiyoloji
KAH vakalarının yaklaşık %5-8'ini oluşturur ve yıllık 1/100,000-200,000 canlı doğum insidansı vardır.
Klinik Tanım
Bozukluk neonatal dönemde tanımlanmazsa, hem dişi hem de erkek çocuklar, hızlı büyüme hızı, hızlı erişkin iskelet olgunlaşması (erişkinlikte kısa boya neden olur) ve cinsel önkoşul ile hızlı bir şekilde doğum sonrası büyümeye maruz kalırlar. Erkeklerin normal görünmesine karşın kızların dış cinsel organlarında ciddi virilizasyon görülür. Her iki cinsiyette de erken dönemde yalancı puberte ve hipertansiyon görülür. Ayrıca adrenal bir kriz için yaşam boyu risk vardır.
Etiyoloji
Hastalığa, 8q21 kromozomunda bulunan CYP11B1 genindeki bir mutasyon neden olur. Steroid 11 beta-hidroksilaz eksikliği, glukokortikoid ve mineralokortikoid öncüllerinin birikmesi nedeniyle kortizol salınımının azalmasına ve hipertansiyona neden olur.
Genetik Danışmanlık
Hastalık otozomal resesif kalıtım gösterir.
Uzman(lar): Pr Juliane LEGER – Son Güncelleme: Ekim 2012
Çeviren: Can Veysel ŞOROĞLU, MSc
Hastalık Tanımı
Glukokortikoid eksikliği, hipergonadotropik hipogonadizm ve ciddi hipokalemik hipertansiyon ile karakterize çok nadir görülen bir konjenital adrenal hiperplazi (KAH).
ORPHA: 90793
• Eş Anlamlısı(ları):
o 17-alfa-hidroksilaz eksikliğine bağlı CAH
o Kombine 17-hidroksilaz / 17,20-liyaz eksikliği
• Prevalans: 1-9 / 1.000.000
• Kalıtım: Otozomal resesif
• Başlangıç yaşı: Bebeklik, Yenidoğan
• ICD-10: E25.0
• OMIM: 202110
• UMLS: C0268285
• MeSH: -
• GARD: 1469
• MedDRA: -
Özet
Epidemiyoloji
Tüm KAH vakalarının yaklaşık %1'ini oluşturur. Prevalans bu nedenle 1/1.000.000 civarındadır.
Klinik Tanım
Hem cinsiyet steroidleri hem de glukokortikoid eksikliği vardır. Yaygın belirtiler arasında erkeklerde cinsel gelişim yetersizliği/düşük virilization, kadınlarda primer amenore ve her iki cinsiyette de pubertal gelişim eksikliği vardır. Genellikle hipokaleminin eşlik ettiği hipertansiyon, bu hastalıkta görülen mineralokortikoid fazlalığı nedeniyle de gelişebilir.
Etiyoloji
Hastalığa, 10q24.3 kromozomunda bulunan CYP17A1 genindeki bir mutasyon neden olur.
Genetik Danışmanlık
Hastalık otozomal resesif kalıtım gösterir.
Uzman(lar): Pr Juliane LEGER – Son Güncelleme: Ekim 2012
Çeviren: Can Veysel ŞOROĞLU, MSc
Hastalık Tanımı
Konjenital adrenal hiperplazi (KAH) 'nin en yaygın şekli olan, dişilerde kuşkulu genitalya ve adrenal yetmezlikle (her iki cinsiyette de), yenidoğan döneminde dehidratasyon ve hipoglisemi ile ortaya çıkan, basit virilizan veya tuz kaybettiren formları olan ve hiperandrojeneminin görüldüğü bir bozukluk (eğer tedavi edilmezse ölümcül olabilir).
ORPHA:90794
- Eş Anlamlısı(ları): Klasik 21-HE KAH
- Prevalans: 1-9 / 100 000
- Kalıtım: Otozomal resesif
- Başlangıç yaşı: Antenatal, yenidoğan
- ICD-10: E25.0
- OMIM: 201910
- UMLS: C2936858
- MeSH: -
- GARD: 12665
- MedDRA: -
Özet
Epidemiyoloji
Prevalans yaklaşık 1/14.000'dir.
Klinik Tanım
Klasik 21-HE KAH iki klinik gruba ayrılabilir: basit virilizan veya tuz kaybettiren (bu terimlere bakınız). Klasik 21-HE KAH'ın klinik bulguları doğum öncesinde veya doğumda gözlenir. Dişilerde kuşkulu genitalya (klitoromegali, ruga ile kısmen kaynaşmış labia major, yaygın ürogenital sinüs) ile ortaya çıkar ve virilizasyonun boyutu normal erkek görünümünden minimal klitoromegaliye kadar değişebilir. Normal uterus ve çeşitli derecelerde anormal vajinal gelişim görülür. Erkeklerde dış genitalya normaldir. KAH'ın tuz kaybettirici formları, aldosteron eksikliği nedeniyle yaşamın ilk birkaç haftasında dehidratasyon ve hipotansiyon semptomlarına yol açar. Hemen tedavi edilmezse, hayati tehlike oluşturabilecek, hiponatremi, hiperkalemi, asidoz ve hipoglisemi gelişebilir. Hiperandrojenemi, kendini hızlanmış büyüme hızı ve iskelet olgunlaşması (erişkinlikte kısa boya yol açar), ileri kemik yaşı, erken çocukluk döneminde erken pubarş ve erken puberte, akne ve hirsutizm, ergenlik döneminde adet problemleri, subfertilite, metabolik bozukluklar ve obezite ile kendini gösterir.
Etiyoloji
Hastalığa, kortizol ve aldosteron üretimini kontrol eden 6p21.3 kromozomu üzerinde bulunan CYP21A2 genindeki bir mutasyon neden olur.
Tanısal Yöntemler
Klasik 21-HE KAH'lı dişilerin teşhisi genellikle kuşkulu genitalya tespiti ile doğumdadır. Amniyotik sıvıda bulunan 17-hidroksi-progesteron (17-HP) seviyelerini ölçerek doğum öncesi KAH fetüslerde teşhis edilebilir. Çoğu Avrupa ülkesindeki ulusal sistematik yenidoğan tarama programları KAH vakalarını teşhis etmektedir.
Ayırıcı Tanı Ayırıcı tanılar arasında diğer KAH formları, polikistik over sendromu (PKOS, bu terimlere bakınız) veya androjen fazlalığı olan diğer hastalıklar yer alır.
Antenatal Tanı
Prenatal test, 10-11. gebelik haftasında koryon villus örneklemesi (CVS) veya 15-18. GH'da amniyosentez ile 17-HP'nin enzim aktivitesi ölçümü ile yapılabilir.
Genetik Danışmanlık Klasik 21-HE KAH, otozomal resesif kalıtım modelindedir ve genetik danışmanlık mümkündür.
Yönetim ve Tedavi
Klasik KAH riski taşıyan dişi fetüsler için deksametazon ile doğum öncesi tedavi uygulanabilir. 9. haftadan önce uygulandığında, dişilerdeki kuşkulu genitalyadan sorumlu aşırı androjen üretimini önler. Doğumdan sonra teşhis konulursa, vajinoplasti ameliyatı genellikle yaşamın ilk yılında yapılır. Normal büyüme ve ergenlik için, adrenal yetmezliğini tedavi etmek ve yüksek androjen hormonu seviyelerini azaltmak için yaşam boyu hormon replasman tedavisi gerekir. Hidrokortizon genellikle çocuklara glukokortikoid (GK) replasman tedavisi olarak (10-15mg / m2 / gün 2 veya 3 doza bölünmüş) ve mineralokortikoid (MK) replasmanı için 9 alfa-fludrokortizon verilir. Doz, hasta stres altında olduğu zamanlarında izlenir ve değiştirilir. Akut adrenal yetmezlik (bu terime bakınız) ve kronik hiperandrojenemiye bağlı diğer komplikasyonların gelişme riski vardır. GK ile aşırı tedavi cushingoid yan etkilere neden olur ve aşırı MK hipertansiyona neden olur. Pediatrik endokrinologlar, cerrahlar, jinekologlar, psikologlar dahil olmak üzere multidisipliner bir ekip tarafından düzenli takip önemlidir.
Prognoz
Uygun tedavi altında hastalar normal bir yaşam beklentisine sahiptir. Uzman(lar): Pr Juliane LEGER – Son Güncelleme: Ekim 2012
Çeviren: Ezgi Gizem Berkay, MD – OCAK 2019
Kaynak sayfa için tıklayınız
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
22q11.2 delesyon sendromu (DS) konjenital malformasyon hastalığına neden olan bir kromozomal anomalidir. Kardiyak hasarlar, palatal anomaliler, yüz dismorfizmi, büyüme gecikmesi ve immune yetmezlik sık karşılaşılan özellikleridir.
ORPHA:567
- Eş Anlamlısı(ları):
- 22q11DS
- CATCH 22
- Cayler kardiyofasiyal sendrom
- Konotronkol anomaly yüz sendromu
- DiGeorge sekansı
- DiGeorge sendromu
- Mikrodelesyon 22q11.2
- Monozomi 22q11
- Sedlackova sendrom
- Shprintzen sendrom
- Takao sendrom
- Velokardiyofasiyal sendrom
- Prevalans: Bilinmiyor
- Kalıtım: Otozomal Dominant ya da bilinmiyor
- Başlangıç yaşı: Neonatal
- ICD-10: D82.1
- OMIM: 188400 192430
- UMLS: C0012236 C0220704 C0431406 C0795907 C2936346 C3266101
- MeSH: D058165
- GARD: 10299
- MedDRA: 10012979 10066430
Özet
Epidemiyoloji
Dünya genelindeki insidansı 1/2000-1/4000 canlı doğum olarak tahmin edilmektedir.
Klinik Tanım
22q11.2 DS, hafif ila şiddetli arasında değişken bir klinik fenotipi göstermektedir. Konjenital kalp defektleri (olguların %77'sinde), trunkus arteriosus, Fallot tetralojisi ve ventriküler septal defekt gibi başlıca konotrunkal malformasyonları içerir. Hastaların %75'inden fazlasında hipernazal konuşma, beslenme ve yutma güçlüğüne yol açabilen palatal anomaliler (örneğin yarık damak, yarık dudak ve damak, veloparingeal yetersizlik) görülür. Gelişimsel gerilik sık gözlenir. Birçok hasta, hafif yüz dismorfizmi (örneğin, malar düzlük, pitoz, hipertelorizm, epikantal kıvrımlar, belirgin nazal kök) ve vertebral anomaliler (örneğin, kelebek vertebraları, hemivertebralar) ile başvurmaktadır. Hastaların %75'inde, çeşitli enfeksiyonlara duyarlı hale getiren timik aplazi / hipoplazi nedeniyle immün yetmezliği vardır. Hastalar ayrıca idiopatik trombositopenik purpura ve juvenil idiyopatik artrit gibi bir otoimmün hastalık geliştirme riski daha yüksektir (bu terimlere bakınız). Neonatal hipokalsemi olguların %50'sinde görülür. Genellikle düzelir ancak herhangi bir yaşta veya bir enfeksiyondan, ameliyattan veya hamilelikten sonra tekrar ortaya çıkabilir. Ek klinik bulgular arasında gastrointestinal anomaliler (intestinal malrotasyon, imperfore anüs), işitme kaybı, renal anomaliler (renal agenezis), dental anomaliler (mine hipoplazisi), öğrenme problemleri ve / veya psikiyatrik bozukluklar (dikkat eksikliği hiperaktivite bozukluğu, şizofreni) sayılabilir. Sendromun kapsadığı klinik fenotiplerin geniş spektrumu daha önce farklı sendromlara (örn., DiGeorge sendromu, velokardiofasiyal sendrom, kardiyofasiyal sendrom) bölünmüştür, ancak şu anda etiyolojik olarak özdeş olduğu bilinmektedir ve 22q11.2 DS olarak adlandırılmaktadır.
Etiyoloji
Vakaların büyük bir kısmında sendrom, 22q11.2 kromozom bölgesindeki düşük kopya sayısı tekrarlarıyla çevrili 3 milyon baz çiftlik (Mb) bir delesyondan kaynaklanmaktadır. Delesyon spermatogenez ya da oogenez sırasında alelik olmayan mayotik rekombinasyon kaynaklıdır. Vakaların %15’inde delesyon 3Mb’lik bölgenin içindedir ve farklı boylarda olabilir. Ayrıca DiGeorge kritik bölgesi içinde atipik delesyonlar da mevcuttur. Bazı delesyonlar kardiyak, paratiroid, timus ve yüz yapı gelişimine katılan TBX1 genini kapsamaktadır. 22q11.2 fenotipinde gözlenen değişken ekspresivitenin diğer 22q11.2 alellindeki ya da diğer kromozomlardaki genetik modifiye edici genlerden kaynaklandığı düşünülmektedir.
Tanı Yöntemleri
Klinik inceleme ve anomaliler (ör: ekokardiyografi ile kardiyak hasarlar, servikal omur X-ışınları ile vertebral anomaliler) gözlenmesi üzerine tanıdan şüphelenilir. Floresan in situ hibridizasyon (FISH), MLPA, cCGH ya da genom boyu SNP mikroarrayleri gibi yöntemlerle 22q11.2 delesyonunun gösterilmesi ile tanı kesinleştirilir
Ayırıcı tanı
Ayırıcı tanı Smith-Lemli-Opitz sendromu, CHARGE sendromu, Alagille sendromu, VATER sendromu, Goldenhar sendromu ve izotretinoin embriyopatisi (bu terimlere bakınız).
Antenatal Tanı
Ailesel vakalarda ve fetal ekokardiyogragi ile ilişkili anomaliler tespit edilmiş gebeliklerde, koriyonik villus öneklemesi ya da amniyonsentez ile prenatal tanı mümkündür. Preimplantasyon genetik tanı uygulanabilir.
Genetik Danışma
De novo vakaların yaklaşık %90’ında delesyonlar görülür. Etkilenmiş bireylerde %50 nüks riski mevcuttur.
İdame ve Tedavi
Tedavi ilişkili anomalilere bağlıdır. Kalp ve/ya da damak operasyonları, konuşma terapisi, nasogastrik besleme, kalsiyum takviyesi ve psikolojik terapiyi içerebilir. Düzenli immünolojik takip gereklidir.
Prognoz
Prognoz değişkendir ve hastalığın şiddetine bağlıdır. İnfant ölümleri görece düşüktür (~%4). Erişkinlerde, diğer erişkin populasyona göre mortalite daha yüksektir
Uzman Hakem(ler): Dr Donna MCDONALD-MCGINN - Dr Elaine ZACKAI – Son Güncellme:Aralık 2012
Çeviren: Özden Hatırnaz Ng, PhD- Ocak 2019
Kaynak sayfa için tıklayınız
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
3-metilkrotonil-CoA karboksilaz eksikliği (3-MCCD), bebeklik döneminde metabolik krizden asemptomatik erişkinlere kadar oldukça değişken bir klinik tablo ile karakterize olan kalıtsal bir lösin metabolizması bozukluğudur.
ORPHA:6
- Eş anlamlısı(ları) : 3-metilkrotonilglisinüre, MCC eksikliği, MCCD
- Prevalans: 1-9 / 100 000
- Kalıtım: Otozomal resesif
- Başlangıç Yaşı: Tüm yaşlar
- ICD-10: C25.0 C25.1 C25.2 C25.7 C25.8
- OMIM: 210200 210210
- UMLS: C0268600
- MeSH: C535308
- GARD: 10954
- MedDRA: -
Özet
Epidemiyoloji
Avrupa'da doğum prevalansının 1/50,000-1/30,000 olduğu tahmin edilmektedir. Tandem kütle spektrometrisine dayanan yenidoğan tarama programlarının başlatılması, bu hastalığın sıklığını ortaya çıkarmıştır ve şimdilerde bazı popülasyonlarda en yaygın organik asitüri olduğu görülmektedir.
Klinik Tanım
3-MCCD'li hastalar değişken asemptomatik ve çoğu zaman çevresel tetikleyici faktörlerle birlikte organik bir asidüri semptomları gösteren küçük bir alt gruba sahip değişken bir klinik fenotipe sahiptir. Şimdi genişletilmiş yenidoğan tarama testleriyle tanı konan birçok yenidoğan asemptomatik kalıyor ve bu hastalığın çok düşük bir klinik penetrasyona sahip olduğunu işaret etmektedir. Semptomatik hastaların çoğu, 2-33 aylık yaşlarda, genellikle küçük bir enfeksiyon, açlık veya protein açısından zengin bir diyetin uygulanmasından sonra akut metabolik krizle ortaya çıkana kadar normal büyüme ve gelişme gösterir. Belirtileri kusma, koma ve apne içerir. Nadiren nörolojik anormallikler (yani metabolik inme, hemiparezi ve ensefalopati), zayıflık, kas hipotoni ve gelişimsel gecikme bildirilmiştir. Metabolik kriz dönemleri arasında hastalar genellikle asemptomatiktir. 3-MCCD'li bazı hastalar yetişkinliğe kadar, zayıflık ve halsizlikle kendini gösteren semptomlar geliştiremezken, diğerleri hiçbir zaman semptom göstermeyebilir.
Etiyoloji
3-MCCD, MCCC1 (3q27.1) veya MCCC2 (5q12-q13) genlerindeki mutasyonlardan kaynaklanmaktadır. Bu iki gen, birlikte lösin katabolik yolundaki dördüncü basamağı katalize eden MCCase alt birim alfa ve MCCase alt birim beta kodlar. Bu genlerdeki mutasyonlar azalmış veya yokmuş 3-MCC aktivitesine yol açar, böylece lösin işleminin toksik yan ürünlerinin birikmesine ve klinik semptomlara neden olmasına izin verir. İkinci bir nadir hastalık genindeki mutasyonlara zarar veren mutasyonlar için homozigosite ile birlikte olmanın, 3-MCCD'nin spesifik olmayan şiddetli fenotipleri ile ilişkili olabileceğine dair kanıtlar ortaya çıkmaktadır.
Tanısal Yöntemler
Tandem kütle spektrometresi kullanılarak yapılan yenidoğan taraması, kan lekelerinde C5-hidroksi asilkarnitin yükselmesi olduğunu göstermektedir. İdrar organik asit analizi, 3-hidroksi izovalerik asit ve 3-metilkromilglisin seviyesini gösterir. Karnitin serum seviyeleri azaltılabilir. Lenfosit ve fibroblast deneyleri, düşük veya eksik 3-MCC aktivitesi gösterirken, diğer karboksilaz enzimleri normal aktiviteye sahiptir. Akut metabolik kriz sırasındaki laboratuvar bulguları arasında metabolik asidoz, hipoglisemi ve bazı durumlarda hafif hiperammonemi vardır. Allellere neden olan iki hastalığı tanımlayan moleküler genetik testler tanıyı doğrular. Yenidoğan tarama programları ABD'de ve bazı Avrupa ülkelerinde mevcuttur.
Ayırıcı Tanı
Ayırıcı tanı, Reye sendromunun yanı sıra çoklu karboksilaz eksikliği gibi diğer organik asitleri de içerir (bu terimlere bakınız).
Antenatal Tanı
Antenatal tanı, hastalığa neden olan mutasyonlara neden olan ailelerde mümkündür.
Genetik Danışmanlık
3-MCCD, otozomal resesif olarak kalıtılır ve genetik danışmanlık önerilir.
Yönetim ve Tedavi
Erken tanı 3-MCCD'nin uygun şekilde yönetilmesinde ve ciddi metabolik kriz riskinin azaltılmasında yardımcı olabilir. Asemptomatik bireylerde tedavi genellikle gerekli değildir. Bazı hastalarda oral L-karnitin takviyesi gerekebilir. Lösin ile sınırlı bir diyet genellikle garanti edilmez. Serbest karnitin konsantrasyonlarının düzenli olarak izlenmesi gibi oruç tutmanın ve diğer çöktürücü gerilmelerin önlenmesi (özellikle bebeklerde ve küçük çocuklarda) önerilmektedir. Akut hastalık sırasında acil bir rejim (IV glikoz ve asidozun düzeltilmesi) önerilmektedir.
Prognoz
Prognoz genellikle iyidir ancak yaşanan semptomların ciddiyetine bağlıdır.
Uzman Hakem(ler): Pr Matthias BAUMGARTNER – Son Güncelleme:Ekim 2014
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
46, XX yumurtalıkta cinsel gelişim bozukluğu (46, XX yumurtalıkta DSD), 46, XX karyotipli bir bireyin histolojik olarak doğrulanmış testis ve yumurtalık dokusu ile karakterize edilir.
ORPHA:2138
- Eş anlamlısı(ları) : 46,XX ovotestiküler DSD
- Prevalans: 1-9 / 100 000
- Kalıtım: Otozomal resesif
- Başlangıç Yaşı: Bebeklik, Yenidoğan
- ICD-10: Q56.0
- OMIM: 400045
- UMLS: C0266361 C2748895
- MeSH: D058165
- GARD: -
- MedDRA: -
Özet
Epidemiyoloji
Tahmini prevalans yaklaşık 1/20.000 doğumdur. Hastalık tüm DSD'nin %3-10'undan daha azını oluşturabilir.
Klinik Tanım
Etkilenen bireylerin yaklaşık %20'si, 5 yaşından önce teşhis edilir. Atipik genital organlara bağlı olarak tanı genellikle yenidoğan döneminde yapılır. Bazıları daha sonra anormal pubertal gelişim ile ortaya çıkar. Belirtiler alt karın ağrısı, jinekomasti, kasık fıtığı, bir inguinoskrotal kitle, kriptorşidizm veya cinsiyet atamasına bağlı olarak amenore/periyodik hematüri. Etkilenen bireylerin çoğu kadın iç genital organına (uterus, hemi uterus veya ilkel uterus) sahiptir. Dış genital organların gelişimi, belirgin kadın genital organından erkek genital organına korid ve hipospadias ile birlikte değişir. Nadiren hastalar normal veya normale yakın bir erkek penisine sahiptir ve bilateral olarak inişli ovotestlere (over ve testis elementleri içeren gonadlar) sahiptir. Gonadal yapılandırmaları değişiklik gösterir. Kısırlık erkeklerde sık görülürken, kadınların doğurganlık için biraz potansiyelleri vardır. Malign gonadal tümörleri nadir görülür (vakaların %3'ünden azı).
Etiyoloji
46, XX ovotestiküler DSD'nin kesin nedeni, vakaların çoğu için açıklığa kavuşturulmamıştır, ancak mozaikizm veya genetik mutasyonlarla ilgili olabilir. Olguların çoğu (% 65) SRY (Y kromozomunun cinsiyet belirleyici bölgesi) -negatiftir. Nadiren, hastalar SRY geninin bir kopyasına sahiptir, SOX9 gen çoğalmasına ya da RSPO1 ya da WNT4 genlerinde, hurda hücreli karsinom ve Serkal sendromuna sırasıyla palmoplantar keratoderma - XX cinsiyet ters - predispozisyonunun bir parçası olarak mutasyonlara sahiptir (bu terimlere bakınız).
Tanısal Yöntemler
Birçok hasta belirgin genital belirsizliğe sahiptir ve doğumda teşhis edilir. DSD'nin değerlendirilmesi ve tanısı karmaşıktır. Mutabakat kuralları, muayene ve tedavi için uzman bir merkeze başvurmanızı önerir. İlk araştırmalar kromozom analizini ve iç üreme organlarını kontrol etmek için bir ultrason taramasını içerir. Hastalığı sonradan gösteren hastalar, cinsel organlarda daha yüksek bir farklılaşmaya sahiptir. Teşhis görüntüleme yöntemleri ve/veya laparoskopi ile dikkatli bir anatomik değerlendirme gerektirir. Biyokimyasal endokrin incelemesi, sitogenetik ve moleküler genetik testler gereklidir. Kesin tanı gonadal histolojiye dayanmaktadır (testis ve over dokusu).
Ayırıcı Tanı
Ayırıcı tanılar, karışık gonadal disgenezi ve 46,XX testis DSD'si dahil diğer DSD'yi içerir (bu terimlere bakın).
Antenatal Tanı
Prenatal tanı atipik genital organların ultrasonda gösterilmesinin ve amniyosentezde bir 46, XX karyotip ortaya çıkmasının ardından mümkün olabilir.
Genetik Danışmanlık
Etkilenen çocukların ailelerine genetik danışmanlık verilmelidir. Tekrarlama riski, bulunan genetik değişikliğin türüne bağlıdır. Çoğunlukla de novo mutasyonları olarak ortaya çıkar.
Yönetim ve Tedavi
Hasta ve aileye psikolojik destek sağlanmalıdır. Diğer tedaviler temel olarak hormon replasmanını içerir. Cinsiyet ataması ve gonadal konfigürasyona bağlı olarak cerrahi tedaviye duyulan ihtiyaç ve zamanlama karmaşıktır. Yönetim gonadektomi ve rekonstrüktif cerrahi risklerini ve yararlarını dengelemeye ihtiyaç duyar.
Prognoz
Hastalar genellikle normal yaşam beklentisine sahiptir.
Uzman Hakem(ler): Pr Faisal AHMED - Dr Angela LUCAS-HERALD - Dr Ruth MCGOWAN - Pr Edward TOBIAS Son
Güncelleme:Ocak 2014
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
46,XX testis cinsiyet gelişimi bozukluğu (46,XX testis DSD) testosteron eksikliği ile birlikte, normalden belirsizliğe dek değişen erkek dış genital organı ile karakterizedir.
ORPHA:393
- Eş anlamlısı(ları) : 46,XX testiküler DSD
- Prevalans: 1-9 / 100 000
- Kalıtım: -
- Başlangıç Yaşı: Yeni doğan,bebeklik
- ICD-10: Q99.1
- OMIM: 278850 300833 400045
- UMLS: C0432475 C2936419
- MeSH: D058531
- GARD: 399
- MedDRA: -
Özet
Epidemiyoloji
Tahmini prevalans 1/20.000 erkek olarak görülür.
Klinik Tanım
Klinik fenotip, aşağıdakileri içeren özelliklerle değişkendir: atipik genital dış normal erkek, Müllerian yapıları olmayan infertiliteye sahip inmemiş testis. Prezentasyonu, SRY geninin varlığına bağlıdır (Y kromozomunun cinsiyet belirleyici bölgesi). SRY pozitif olgular (%80-90) genellikle kısa boylu, normal pubik kıl ve penil büyüklüğü olan ergenlik sonrası ortaya çıkan, ancak küçük testisler, jinekomasti ve azoospermiye bağlı kısırlığı olan normal erkeklerdir. Kriptorşidizm ve hipospadias da bildirilmiştir. Cinsiyet rolü ve kimliği hakkında genellikle endişeler yoktur. SRY negatif bireyler (%10-20) genellikle doğumda penoskrotal hipospadias ve kriptorşidizm gibi özelliklerle görülür. Erkek hipogonadizmi nedeniyle uzun vadeli komplikasyonlar şunlardır: düşük libido, erektil işlev bozukluğu, sekonder cinsel özelliklerin azalması, osteopeni ve depresyon.
Etiyoloji Çoğu hastada, hastalığa SRYin bir karyotipik XX genomu içeren küçük bir Y-kromozom parçasının varlığı neden olur. Normal bir SRY alelinin bir X kromozomuna translokasyonu yaygın bir nedendir, ancak SRY-negatif vakalarda, diğer genler örn. SOX9, SOX3, RSPO1 ve WNT4 de ilişkilidir.
Tanısal Yöntemler
Tanı hipergonadotrofik hipogonadizm ve sitogenetik testi gösteren klinik bulgulara ve endokrin testlerine dayanmaktadır. Floresan in situ hibridizasyon (FISH) veya polimeraz zincir reaksiyonu (PCR) teknikleri SRY genini tespit edebilir.
Ayırıcı Tanı
Başlıca ayırıcı tanılar 45,X/46,XY karışık gonadal disgenezi, 47,XXY Klinefelter sendromu, 46,XX ovotestiküler DSD ve cinsiyet kromozom mozaikleridir. Nadiren diğerleri arasında derinin skuamöz hücreli karsinomalı palmoplantar keratoderma ve XX cinsi tersine çevirme (RSPO1 gen mutasyonları nedeniyle) ve lineer cilt defekti (MIDAS) sendromlu mikroftalmi (MIDAS) sendromu bulunur (bu terimlere bakınız).
Antenatal Tanı
SRY pozitif olan riskli gebeliklerde doğum öncesi testler 46,XX testis DSD'si mümkündür.
Genetik Danışmanlık
SRY pozitif olgular genellikle kalıtsal değildir, çünkü SRY geni X kromozomunda mevcut olduğunda, neredeyse her zaman kısırlığa neden olur. Penetrans %100 kadar yüksek olabilir. SRY-negatif vakaların çoğunda, diğer genlerde mutasyonlar tespit edilmedikçe, örneğin; RSPO1 veya WNT4 (her ikisi de resesif kalıtım gösteren), kalıtımın çeşidi bilinmez.
Yönetim ve Tedavi
Tedavinin temeli hormonal dengesizliği düzeltmek, jinekomastiyi önlemek ve erkek sekonder cinsiyet özelliklerinin gelişimini sağlamak için testosteron replasman tedavisidir. Hipergonadotrofik hipogonadizm yetişkinlikten önce nadirdir. Bazı vakalarda küçültme mamoplastisi düşünülebilir. Psikolojik destek ve yönlendirme ile destek servisi önerilmelidir.
Prognoz
Erkek hipogonadizminin yönetimi komplikasyonları azaltır. Etkilenen hastalar tipik olarak kısırdır. Tümorigenik risk düşüktür.
Uzman Hakem(ler): Pr Faisal AHMED - Dr Angela LUCAS-HERALD - Dr Ruth MCGOWAN - Pr Edward TOBIAS– Son
Güncelleme:Aralık 2013
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
47, XYY sendromu, erkeklerin ek Y kromozomu aldıkları bir cinsiyet kromozomu anöploididir ve klinik olarak çocukluk çağında uzun boy ile belirgin , makrosefali, yüz özellikleri (hafif hipertelorizm, alçak kulaklar, hafif düz malar bölgesi), konuşma gecikmesi ve, sosyal ve duygusal zorluklarda risk artışı, dikkat eksikliği hiperaktif bozukluk ve otistik spektrum bozukluğu ile karakterize edilir.
ORPHA:8
- Eş anlamlısı(ları) : Çift Y Sendromu, XYY Sendromu, Y Dizomi
- Prevalans: 1-9 / 100 000
- Kalıtım: Uygulanabilir veya bilinmeyen
- Başlangıç Yaşı: Tüm yaşlar
- ICD-10: Q98.5
- OMIM: -
- UMLS: C0043379 C3266843
- MeSH: C535317 D014997
- GARD: 5674
- MedDRA: 10056894
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
48, XXXY sendromu, erkeklerde iki ekstra X kromozomunun varlığı ile karakterize edilen anöploidik tipte bir kromozomal anomaliyi temsil eder.
ORPHA:96263
- Eş anlamlısı(ları) : -
- Prevalans: 1-9/100.000
- Kalıtım: Bilinmiyor ya da uygulanabilir değil
- Başlangıç Yaşı: Yeni doğan, bebeklik
- ICD-10: Q98.1
- OMIM: -
- UMLS: C0265498
- MeSH: -
- GARD: 5676
- MedDRA: 10048228
Özet
Epidemiyoloji
Yıllık 1/50.000 erkek doğum insidansı vardır.
Klinik Tanım
48,XXXY sendromu, orta derecede entelektüel eksiklik (ortalama IQ:50), daha belirgin genital hipoplazi (mikroorganizma, mikropenis, skrotumun hipoplazisi) ve daha sık gözlenen yüz dismorfikliği (yassı burun, epicanthus, prognatizm, kısa boyun, hipertelorizm, yüz asimetrisi) ile Klinefelter sendromundan farklıdır. Diğer dismorfik özellikler (beşinci parmağın klinodaktili, koksa valga, vb.) bu sendromla sıklıkla ilişkilidir. Genital anomalilere (kriptorşidizm) ve jinekomastiye ek olarak konjenital iskelet malformasyonları (kifoskolyoz, radioulnar sinostoz, epifiz displazisi) de mevcut olabilir. Yaşla birlikte, artropatiler, şişmanlık, davranışsal problemler (hiperaktivite, huzursuzluk, kaygı, olgunluk, pasiflik, öfke, iletişim ve sosyalleşme sorunları) ve dil geriliği gibi diğer belirtiler de ortaya çıkabilir.
Etiyoloji
En muhtemel etiyoloji, homolog kromozomların (ilk mayotik bölünme sırasında) homolog kromozomların (ikinci mayotik bölünme sırasındaki) veya ebeveyn üreme hücrelerinde kardeş olmayan ayrışmasıdır. Bu sendromun gelişmesinden sorumlu veya lehine bilinen bir faktör yoktur.
Tanısal Yöntemler Metafaz karyotipi klinik bir tanının onayına izin verir. Diğer poligonozomlarla mozaiklik nadir değildir (48, XXXY / 48, XXYY / 49, XXXXY).
Ayırıcı Tanı Ayırıcı tanı Klinefelter sendromu (47,XXY), 48, XXYY sendromu ve 49,XXXXY sendromu (bu terimlere bakınız) gibi diğer anöploidleri içerir.
Antenatal Tanı Antenatal tanı amniyosentez ile mümkündür.
Genetik Danışmanlık 48, XXXY vakaları sporadik olması nedeniyle tekrarlama riski çok düşüktür.
Yönetim ve Tedavi
Tedavinin multidisipliner bir ekip tarafından ele alınması gerekir ve kalp ve iskelet malformasyonlarının tedavisini, duyusal, nörolojik, hormonal (testosteron bazlı hormon tedavisi), metabolik (obeziteyi denetleyen), psikolojik ve psikiyatrik bakımı ve diş takibini içerir.
Prognoz
Hastalar temel olarak normal yaşam süresine sahiptir, ancak özellikle endokrin ve bulaşıcı problemleri için düzenli tıbbi ziyaretlere katılmaları ve düzenli psikiyatrik izlenmeleri gerekir. Uzman Hakem(ler): Dr Carole CORSINI - Pr Pierre SARDA – Son Güncelleme:Mayıs 2011
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
Genetik olarak, erkeklerde ekstra bir X ve Y kromozomunun varlığı ve klinik olarak uzun boyla, kısırlık, kısırlık ve yetersiz testosteron üretimi ile ilişkili işlevsiz testlerin varlığı , bilişsel, duygusal ve sosyal işlev bozukluğu ile ilişkili işlevsiz testlerin ve gelişimsel gecikme ve artmış konjenital malformasyon riski ile karakterize edilen nadir bir cinsiyet kromozomu anomalisi hastalığıdır.
ORPHA:10
- Eş anlamlısı(ları) : -
- Prevalans: Bilinmiyor
- Kalıtım: Uygulanabilir değil ya da bilinmiyor
- Başlangıç Yaşı: Yenidoğan, bebeklik, çocukluk çağı, ergenlik
- ICD-10: Q98.8
- OMIM: -
- UMLS: C2936741
- MeSH: D007713
- GARD: 5677
- MedDRA: 10048230
Özet
Epidemiyoloji
Tahmini prevalans 1/18.000 ila 1/50.000 erkek doğumu arasındadır.
Klinik Tanım
Görünüm, genellikle hafif dismorfik yüz plajiyofali ve düz ayakların eşlik ettiği hipotoni ve global gelişim gecikmesi olan bebeklik döneminde veya erken çocukluk dönemindedir. Ergenlik çağında başlayan ve yetişkinlik döneminde devam eden testis hipogonadizmi neredeyse evrenseldir. Kriptorşidizm, mikropenis ve jinekomasti zaman zaman rapor edilmiştir. Genel olarak, bilişsel yetenekler, zihinsel engelli aralığında tam ölçekli IQ olan erkeklerin yaklaşık 1/3'ü ile sınırda (70-80) arasında olma eğilimindedir. Belirgin derecede düşük sözel akıl yürütme becerileri sıklıkla mevcuttur. IQ'ya göre, iletişim, sosyal beceriler, öz-bakım ve öz-yönelimdeki ortak açıklarla birlikte, adaptif işlevsellik önemli ölçüde bozulur. Önemli diş problemleri (~%90), titreme (~ yetişkinlerin%60'ı), astım / alerji (~% 60), iskelet anomalileri (kulüp ayağı, radioulnar synostosis, cubitus varus ile belirgin dirsekler) skolyoz ve kifoz, tip 2 diyabet (yetişkinlikte ~%20), tromboz (~%18), epilepsi (~%15), şaşılık (~%15), gastrointestinal problemler (besleme intoleransı, reflü, kabızlık), konjenital kalp defektleri ve böbrek anormallikleri gibi bir çok tıbbi durumla sıklıkla ilişkilidir. Spesifik olmayan dismorfik özellikler epikantal kıvrımları, hipertelorizmi ve klinodaktili içerebilir. Davranış örüntüleri, ruhsal kararsızlık, kaygı, obsesif-kompulsif davranışlar ve duygusal olgunluğa ek olarak, dikkat eksikliği / hiperaktivite bozukluğu ve otizm spektrum bozukluğu özelliklerini içerebilir.
Etiyoloji
48,XXYY sendromu, spermatogenez sırasında cinsiyet kromozomlarının bir bozulma olayından veya daha az sıklıkla, hücre bölünmesi sırasında zigotik sonrası mitotik bozulmadan kaynaklanır. Bilinen herhangi bir predispozan faktör yoktur.
Tanısal Yöntemler
Tanı genellikle çocukluk çağında fiziksel ve/veya gelişimsel kaygıların değerlendirilmesi sırasında belirlenir. Tanı standart karyotip veya kromozomal mikroarray ile doğrulanır.
Ayırıcı Tanı
Ana ayırıcı tanılar 47,XXY sendromunu; 48,XXXY sendromunu; 49,XXXXY sendromunu; 45,X/46,XY mozaiklik ve 46,XX erkekleri içerir. Diğer olası örtüşen koşullar arasında Fragile X, Jacob, Prader-Willi, Soto, Börjeson-Forssman Lehman, Weaver ve Cohen sendromları bulunur.
Antenatal Tanı
Hücresiz fetal DNA ile invaziv olmayan prenatal tarama, 48,XXYY ile fetüs tanımlayabilir, ancak bu test teşhis amaçlı değildir. Antenatal tanı, amniyosentez veya koryon villus örneklemesi ile mümkündür.
Genetik Danışmanlık
48,XXYY, tipik olarak <%1'lik bir tahmini nüksetme riski olan sporadik bir anöploidi olayı nedeniyledir. Genetik danışma potansiyel fiziksel, tıbbi, gelişimsel ve psikolojik özelliklerin gözden geçirilmesini içermelidir.
Yönetim ve Tedavi
Kapsamlı disiplinlerarası bakım, ilişkili gelişimsel, tıbbi ve psikolojik koşulları değerlendirmek ve yönetmek için önemlidir. Konjenital defektleri değerlendirmek için ayrıntılı bir fizik muayene, renal ultrason ve ekokardiyografi yapılmalıdır. Görme / işitme taramaları ve rutin diş bakımı yaşam boyu önemlidir. Astım ve epilepsi gibi komorbiditeler, 48,XXYY olmayan bireylerde olduğu gibi teşhis ve tedavi edilmelidir. Ergenlik muayeneleri ve serum hormon profilleri, 10 yaş civarında başlayarak izlenmeli ve hipogonadizm için testosteron takviyesi düşünülmelidir. Ergenlikten itibaren yıllık olarak hiperlipidemi, diyabet ve otoimmün tiroid hastalığı taraması önerilmektedir. Kapsamlı, disiplinlerarası nörogelişimsel ve davranışsal değerlendirme çocukluk boyunca garanti altına alınmıştır. Psikolojik işlevselliği (duygusal ve davranışsal bozukluklar dahil) ve konuşma/dil, motor ve kişisel bakım becerilerini hedefleyen müdahaleler kanıta dayalı ve bireyselleştirilmelidir. Okul ve toplum temelli destek ve hizmetler, tedavi planının bir parçasını oluşturur.
Prognoz
Tedavi olmasa da, ilişkili tıbbi ve psikiyatrik durumlar için uygun tedaviyle yaşam beklentisi normaldir. Sağlıkla ilgili yaşam kalitesi ve bağımsızlık derecesi, semptomların ciddiyetine ve uygun sağlık hizmeti ve destek hizmetlerine erişimine bağlı olarak değişir.
Uzman Hakem(ler): Catherine BUCHANAN - Dr Shanlee DAVIS - Dr Susan HOWELL - Dr Nicole TARTAGLIA - Dr Adrianne VILLAGOMEZ – Son Güncelleme:Ocak 2019
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
49,XXXXY sendromu, erkeklerde üç ekstra X kromozomunun varlığı ile karakterize edilen anöploidik bir kromozomal anomaliyi temsil eder.
ORPHA:96264
- Eş anlamlısı(ları) : -
- Prevalans: Bilinmiyor
- Kalıtım: Bilinmiyor ya da uygulanabilir değil
- Başlangıç Yaşı: Çocukluk çağı
- ICD-10: Q98.1
- OMIM: -
- UMLS: C0265499
- MeSH: -
- GARD: 5679
- MedDRA: -
Özet
Epidemiyoloji
Yıllık 1/85.000 ila 1/100.000 erkek doğum insidansına sahiptir.
Klinik Tanım
49,XXXXY sendromu, çocukluk çağında sıklıkla hemen göze çarpmayan bir entelektüel eksiklikle beraber değişken IQ ile ama yaşla birlikte ilerleyen (orta ila şiddetli) bir bozulma (IQ 70 ile 20 arasında değişen) ile Klinefelter sendromundan farklılık gösterir, çoğu zaman zaten uterusda ve doğumdan sonra yüzdelik üçün altında, bazen de küçük bir boy ve büyüme hormonu eksikliği ile görülebilir. Hipogonadizm, mikropenis, mikroorganizma, skrotumun hipoplazisi ve kriptorşidizm ile şiddetlidir. Jinekomasti nadirdir. Olağanüstü yüz dismorfikliği (hipertelorizm, depresif burun eğriliğine sahip büyük düz burun, artmış palpebral fissürler, epikantus, prognatizm, katlanmış kulaklar, kısa boyun) ve beşinci parmağın diğer dismorfik karakteristikleri (kubitus valgus, düz ayaklar, beşinci parmağın klinoktilisi, eklem laksisi)) sık görünür. Konjenital kalp defektleri (arteriyel kanal) yanı sıra iskelet defektleri (radyulnar sinostoz, epifizeal displazi, coxa valga, kifoscoliosis, kalça ve diz dislokasyonu), serebral (hipoplastik korpus kallosum, arenefali) ve renal defop (renal defop). Aksiyal hipotoni çocuklarda sıklıkla görülür. Yaşla birlikte, entelektüel eksiklik kötüleşir ve dil gelişiminde büyük bir gecikmeyle ilişkilidir. Şaşılık ya da görme keskinliğine neden olabilecek şiddetli ve ilerleyici miyop oluşabilir. Utangaçlık gibi davranışsal problemler de ortaya çıkabilir.
Etiyoloji
Etiyoloji, maternal germ hücrelerinde homolog kromozomların (ilk mayotik bölüm sırasında) veya kardeş kromatitlerin (ikinci mayotik bölüm sırasında) ayrılmamasıdır. Bu sendromun ortaya çıkmasından sorumlu veya bilinen herhangi bir faktör yoktur.
Tanısal Yöntemler Metafaz karyotipi klinik bir tanının doğrulanmasına izin verir.
Antenatal Tanı
Antenatal tanı, çok çeşitli koşullar için bir amniyosentez taraması ile mümkündür.
Genetik Danışmanlık
49,XXXXY vakalarının sporadik olması nedeniyle tekrarlama riski çok düşüktür.
Yönetim ve Tedavi
Yönetimin multidisipliner bir ekip tarafından ele alınması gerekir ve kalp ve iskelet kusurlarının tedavisi, psikomotor gelişimin fizyoterapi ile izlenmesi, psikomotoriklik, konuşma terapisi, ortopedik ve duyusal yönetim (oftalmolojik muayene), nörolojik, hormonal (gerekirse) ve psikolojik bakım ve düzenli diş takibi tedavilerini içermelidir.
Prognoz
Hastalar normal yaşam süresi beklenir ancak düzenli tıbbi ziyaretlere katılmaları gerekecektir. Uzman Hakem(ler): Dr Carole CORSINI - Pr Pierre SARDA– Son Güncelleme:Mayıs 2011
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
ORPHA:240839
- Eş anlamlısı(ları) : -
- Prevalans: Bilinmiyor
- Kalıtım: -
- Başlangıç Yaşı: Tüm yaşlar
- ICD-10: -
- OMIM: -
- UMLS: -
- MeSH: -
- GARD: -
- MedDRA: -
Özet
Bu terim farmakogenetik test için bir endikasyonu gösterir.
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
8p inversiyon duplikasyon/delesyon [invdupdel (8p)] sendromu, klinik olarak hafif ila şiddetli entelektüel eksiklik, ciddi gelişimsel gecikme (psikomotor ve konuşma gelişimi), zaman içinde ilerleyici hipertoni ve ağır ortopedik problemler geliştirme eğilimi gösteren hipotoni, minör yüz anomalileri ve korpus kallozumun agenezisi ile karakterize, nadir görülen bir kromozomal anomalisidir
ORPHA:96092
- Eş anlamlısı(ları) : Invdupdel(8p), İnversiyon 8p Duplikasyon/Delesyon Sendromu
- Prevalans: 1-9 / 100 000
- Kalıtım:Bilinmiyor ya da uygulanabilir değil
- Başlangıç Yaşı: Yenidoğan, bebeklik
- ICD-10: Q99.8
- OMIM: -
- UMLS: -
- MeSH: -
- GARD: -
- MedDRA: -
Özet
Epidemiyoloji
Bilinen yaklaşık 50 vaka raporlanmıştır. Prevalansın 1/22.000 ila 1/30.000 doğumda bir olduğu tahmin edilmektedir.
Klinik Tanım
Yaygın olarak görülen klinik belirtiler arasında gelişimsel gecikme, hafif ila şiddetli bilişsel eksiklik derecesi, anlamlı konuşma ve dil eksikliği veya gecikmesi ve ciddi bir psikomotor gecikmeye katkıda bulunan hipotoni vardır. Invdupdel (8p) olan çocukların çoğunun sözlü olmamakla birlikte mutlu tabiatlı, sosyal ve iletişimsel olduğu bildirilmiştir, ancak bazıları dikkat eksikliği, dürtüsellik ve hiperaktivite gösterebilir. Invdupdel (8p) olan bireylerin yüzde otuz ila elli, çok hafif ila ciddi otistik olan bireyler arasında büyük ölçüde değişen otizmi vardır. Çocukluk çağında daha belirgin olan yüz dismorfikliği, hemen göze çarpmaz ve sıklıkla belirgin bir alın, geçici kellik, korunan burun delikleri, alt dudaktan çıkma, büyük ağız ve kulaklar ve kısa bir boyun içerir. Invdupdelli (8p) yetişkinler normalden çok uzun boyludur ve ilerleyici hipertoni, spastik kuadripleji ve sözleşmeli eklemler ve skolyoz gibi ciddi ortopedik problemler geliştirme eğilimindedir. Kardiyak defektler, göz anormallikleri, üriner sistem anomalileri, erken ergenlik, yüksek damak ve anormal diş gelişimi, ayrıca ekstremite ve çıkık kalça anomalileri bildirilmiştir.
Etiyoloji
- kromozomun kısa kolunun bir terminal delesyonuyla beraber ters bir kopyalama ile çoğunlukla ya D8S349'dan bir pter delesyon ile sentromerden D8S552'ye ters bir kopyalama veya 8p11.2 veya 8p21'den D8S552'ye ters çevrilmiş bir kopyalama ile gerçekleşir. D8349. 8p delesyon işleminin klinik tabloya girmesi, 8p inversiyon çoğaltma yeniden düzenlemesinden daha az önemli görünmektedir. Bugüne kadar, tüm invdupdel (8p) de novo meydana gelmiştir.
Tanısal Yöntemler
Tanı, beyin manyetik rezonans görüntülemede (MRG) korpus kallozumun klinik bulgularına ve agenezisine dayanır ve kromozomal analize yol açar. Delesyon işleminin genetik karakterizasyonu için moleküler teknikler kullanılabilir (FISH, MLPA, CGH dizisi).
Ayırıcı Tanı
Ayırıcı tanı, özellikle 8p21-p22 replikasyonuna sahip olanlar olmak üzere, Trizomi 8p (bu terime bakınız) gibi diğer çoklu konjenital anomalileri/entelektüel eksiklik sendromlarını içerir.
Antenatal Tanı
Antenatal tanı, fetal anormalliklerin (ör. Korpus kallozum agenezisi) ekografik olarak saptanmasına ve amniyosentez veya koryon villus örneklemesinden sonra sitogenetik analizine dayanır.
Genetik Danışmanlık
Genetik danışmanlık tavsiye edilir. Invdupdel (8p) düzenlemeleri de novo'da gerçekleşir. Bununla birlikte, ebeveynler, nadir durumlarda yavrularında daha karmaşık işgal (8p) düzenlemesine yol açabilecek olan 8p23.1 segmentini (yaygınlık 1/4 ila 1/5) içeren zararsız bir ortak inversiyon taşıyabilir.
Yönetim ve Tedavi
Spesifik bir tıbbi tedavi yoktur. Erken yaşta fizyoterapinin yanı sıra mesleki terapi ve konuşma terapisi önerilmektedir. Bazı hastalar müzik terapisinden yararlanır. Hiçbir brüt ortopedik komplikasyon kaydedilmemiştir. Ancak düzenli takip gereklidir.
Prognoz
Yaşam beklentisi konusunda bir rapor bulunmamaktadır. Invdupdel (8p) bireylerin çoğunluğunun ömür boyu tam zamanlı bakıma ihtiyacı olacaktır. Spastik kuadripleji, yaşla yavaş ilerleyici olabilir.
Uzman Hakem(ler): Pr Jean-Pierre FRYNS– Son Güncelleme: Ocak 2013
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
8p23.1 duplikasyon sendromu, temel olarak hafif ila orta dereceli gelişimsel gecikme, entelektüel yetmezlik, hafif yüz dismorfizmi (çıkık alın, kemerli kaşlar, geniş burun köprüsü, kalkık burunlar, yarık dudak ve/veya damak) ve konjenital kalp anomalileri (örneğin, atriyoventriküler septal defekt) dahil, oldukça değişken bir fenotip ile kromozom 8'in kısa kolunun kısmen çoğaltılmasından kaynaklanan nadir bir kromozomal anomali sendromudur. Diğer bildirilen özellikler arasında makrosefali, davranışsal anormallikler (örneğin dikkat eksikliği bozukluğu), nöbetler, hipotoni ve oküler ve dijital anomaliler (poli/sindaktil) bulunur.
ORPHA:251076
- Eş anlamlısı(ları) : dup(8)(p23.1p23.1), trizomi 8p23.1
- Prevalans: 1-9 / 100 000
- Kalıtım: Bilinmiyor ve uygulanabilir değil
- Başlangıç Yaşı: Bebeklik, yenidoğan
- ICD-10: E71.1
- OMIM: -
- UMLS: -
- MeSH: -
- GARD: 10304
- MedDRA: -
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
Yüz, uzuvlar ve genital özellikler ve orantısız bir akromelik kısa boy ile karakterize nadir bir gelişimsel bozukluktur.
ORPHA:915
- Eş anlamlısı(ları) : Aarskog sendromu, Fasiyodijitogenital sendrom, Fasiyogenital displazi
- Prevalans: Bilinmiyor
- Kalıtım: Otozomal resesif, Otozomal Dominant ya da X’e bağlı resesif
- Başlangıç Yaşı: Çocukluk çağı
- ICD-10: Q87.1
- OMIM: 100050 305400
- UMLS: C0175701
- MeSH: -
- GARD: 4775
- MedDRA: 10067148
Özet
Epidemiyoloji
AAS prevalansı bilinmemekle birlikte, literatürdeki 1970'deki ilk tanımından bu yana 100'den az vaka bildirilmiştir. Bununla birlikte, prevalans tahminlerinin 1/25,000 civarında olduğu düşünülmektedir. Dünya çapında moleküler olarak kanıtlanmış yaklaşık 40 vaka yayınlanmaktadır.
Klinik Tanım
AAS ağırlıklı olarak erkekleri ilgilendirir. Yüz özellikleri, hem dişi taşıyıcılarda gözlenen hem de alt taraftaki palpebral fissürler, geniş burun köprüsü, önlenmiş burun delikleri, alçak ayarlanmış ve çıkıntılı kulaklar, ve hipertelorizmini içerir. AAS hastalarında kısa ve geniş el ve ayaklar, parmaklar arası perde (ağ), klinodaktili ve proksimal interfalangeal eklemlerin hiperekstansiyonu ve distal interfalangeal eklemlerde fleksiyonun parmakların kuğu boynu deformitesine neden olduğu görülmektedir. Büyüklük doğumda genellikle normaldir, ancak bebeklik döneminde ve çocuklukta büyüme yavaştır, bu da ergenliğe kadar kısa boylanmaya yol açar, bu genellikle geciktirilir. Genelde, geç gençlerde bir büyüme hamlesi, orta derecede kısa bir boyuta neden olur. Genital anomaliler, kriptorşidizm, makroorşidizm, şal skrotum ve daha nadiren hipospadias içerebilir. Doğurganlık normaldir. Dişi taşıyıcılar, yalnızca hipertelorizm olan ince bir fenotipe sahip olabilir. Hastalar, genellikle erken çocukluk dönemine sınırlı olan öğrenme ve davranışsal engelli bir nörogelişimsel fenotip sunabilir. Varsa, zihinsel bozukluk nadiren şiddetlidir.
Etiyoloji
Klinik ve genetik olarak heterojen olmasına rağmen, hastalığın en iyi karakterize edilen formu FGD1 genindeki mutasyonlar (fasiyogenital displazi 1 geni; Xp11.21) 'dir. Ailelerin çoğu genetik nedenini henüz belirlemediğinden, diğer gen (ler) de rol oynayabilir.
Tanısal Yöntemler
Klinik tanı, fiziki muayeneye ve en belirgin klinik özelliklerin tanınmasına dayanır. FGD1 geninin analizine dayanan moleküler genetik, tanıyı doğrulayabilir.
Ayırıcı Tanı
Moleküler tanı kesin olmadığında, Noonan sendromu, SHORT sendromu, psödohipoparatiroidi ve Robinow sendromu da dahil olmak üzere, ayırıcı tanı için tüm olası seçenekler düşünülmelidir (bu terimlere bakınız).
Antenatal Tanı
Artmış risk altındaki gebeliklerde , ailede hastalığa neden olan mutasyon bilindiğinde, doğum öncesi tanı teknik olarak mümkündür (mutasyonların çoğu aileye özgüdür). Bununla birlikte, doğum öncesi testlerin sıklıkla istenmesi olası değildir, çünkü genellikle fiziksel bulgular hafif olabilir ve klinik heterojenite aynı ailede bile fenotip tahminini zorlaştırır.
Genetik Danışmanlık
AAS, X'e bağlı bir hastalıktır, ancak otozomal dominant ve otozomal resesif yayınlar da rapor edilmiştir. Bu nedenle genetik danışma hastanın aile geçmişinin derinlemesine araştırılmasını gerektirir.
Yönetim ve Tedavi
AAS için iyileştirici bir tedavi yoktur. Çocukluk çağında büyüme hormonu uygulamasının ön sonuçlarının önemli bir etki göstermediği görülmektedir. Öğrenme problemleri ve dikkat eksikliği ve hiperaktivite bozukluğu (DEHB), nöropsikiyatrik bir müdahale gerektirebilir.
Prognoz
Hastaların çoğu iyi bir prognoz gösterir. Tipik olarak, yaşa bağlı zihinsel durum gelişimi ile yetişkinlikte iyi bir evrime sahiptirler.
Uzman Hakem(ler): Dr Alfredo ORRICO– Son Güncelleme :Ekim 2012
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
ACTH'nin anormal üretiminin neden olduğu endojen Cushing sendromu (CS), vakaların %80'inde bir hipofiz adenomunun (Cushing hastalığı, CD) ve ektopik ACTH sekresyonuna göre vakaların %20'sinde adrenokortikotropik hormonun (ACTH) bir ekstrahipofiz tümörü (akciğerlerden veya daha az yaygın timus, pankreas, adrenal bez veya tiroid kaynaklı vakaların %50'sinde) veya çok nadiren ACTH ve kortikotrofin salgılayan hormonu ( CRH) salgılayan bir tümör nedeniyle aşırı miktarda kullanılmasına neden oldu (EAS'ye bağlı CS).
ORPHA: 99892
- Eş anlamlısı(ları) : ACTH'ye bağımlı CS, Adrenokortikotropik hormon bağımlı Cushing sendromu, Kortikotropin-bağımlı Cushing sendromu
- Prevalans: Bilinmiyor
- Kalıtım: -
- Başlangıç Yaşı: -
- ICD-10: E24.0
- OMIM: -
- UMLS: -
- MeSH: -
- GARD:-
- MedDRA: -
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
Olguların % 80'inden fazlasında tibia ön yüzünden ortaya çıkan(tibial diafiz), nadir, primer düşük dereceli malign kemik tümörüdür. Vakaların çoğu semptomatiktir, ağrı, şişlik, eğrilik deformasyonu veya patolojik kırılma ile ortaya çıkar. Özellikle akciğerlere metastaz görülebilir.
ORPHA:55881
- Eş Anlamlısı(ları):
- Uzun Kemiklerin Adamantinoması
- Prevalans: Bilinmiyor
- Kalıtım: Uygulanamaz
- Başlangıç yaşı: Adölesan, Çocukluk, Yetişkin
- ICD-10: C40.2
- OMIM: 102660
- UMLS: C1367554
- MeSH: D050398
- GARD:-
- MedDRA: -
Özet
Epidemiyoloji
AD, tüm primer malign kemik tümörlerinin %0,1 ila %0,5'ini temsil eder. Avrupa'da yaşam boyunca sıklığının 1/900.000 olduğu tahmin edilmektedir. Erkekler daha sık etkilenir ve ikinci durumda tümör daha agresiftir.
Klinik Tanım
AD, geniş yaş aralığında (2-86 yaş) bireyleri etkiler, ancak %75'i ikinci ve üçüncü dekad yetişkin iskeletlerinde görülür. AD'nin sinsi bir başlangıcı vardır ve tipik bulgu tibianın ön tarafında yavaş büyüyen ve ağrısız bir şişliktir. Lokalize ağrı, patolojik kırık ve kemik deformitesi, hastanın tıbbi yardım almasına neden olan diğer özelliklerdir. Tümörler, tibial anterior korteksin orta üçte birinde bulunur, ancak tibia dışındaki kemiklerin de tutulduğu bildirilmiştir (humerus (%6), ulna (%4), fibula (%3), femur (%3) ve radius (%1)). Nadir durumlarda, tümör kaburga, pelvis, omurga, el ve ayak bileklerinde bulunabilir. Birden fazla konumda veya sadece korteks içinde gözlenebilir.
Etiyoloji
AD'nin etiyolojisi bilinmemektedir ve bugüne kadar, bilinen herhangi bir genetik mutasyon tanımlanmamıştır. Bir erkek çocukta tibial AD'den bir akciğer metastazında translokasyon t(7;13) (q32;q14) bildirilmiştir. Ancak bu translokasyon sağlıklı babasında da bulunmuştur.
Tanısal Yöntemler
Konvansiyonel radyografide, erken evre AD'de, periosteal olmayan kortikal belirgin bir berraklık olarak görülür. Kortikal kalınlaşma, kırıktan sonra görülebilmesine ve yumuşak doku kitlesinin varlığına rağmen nadir görülen bulgulardır. Bu evrede tümörlerin tespiti nadirdir ve genellikle tesadüfidir, çoğu hasta daha ileri evrelerde teşhis edilir. Tipik tümörler, karakteristik bir 'sabun köpüğü benzeri' görünüme sahip, çok damarlı kistik/sklerotik lezyonlardır. Bazı durumlarda, korteks sonunda bozulabilir ve yumuşak bir doku bileşeni bulunabilir. Lezyonlar çoğunlukla eksantriktir, genişler ve kortikal yerleşimlidir. AD, T-1 ağırlıklı MRG'de düşük sinyal yoğunluğunu ve T-2 ağırlıklı görüntülerde yüksek sinyal yoğunluğunu gösterir. Histopatolojik terimlerle AD, 2 farklılaştırıcı düzende sonuçlanan osteofibröz doku içerisinde değişken oranlarda birbirleriyle karışabilen epitelyal hücrelerin mevcudiyeti ile karakterize edilir: orta derecede malignite potansiyeli olan osteofibröz displaziye benzer AD ve epitelyal komponent ağırlıklı ve düşük dereceli maligniteli klasik AD.
Ayırıcı Tanı
AD için temel ayırıcı tanı kondrosarkomun fibröz displazisidir, ancak buna Langerhans hücreli histiyositoz, hemanjioendotelyoma (bu terimlere bakınız), fibroma (kemikleşmenin yanı sıra kemikleşmeyen) ve kemik kisti dahil edilmemelidir.
Yönetim ve Tedavi
Kesin iyileşme, ancak geniş rezeksiyon sınırlarına sahip geniş total rezeksiyondan sonra sağlanabilir. Kemoterapi ve radyoterapi AD'lerin tedavisinde rol oynamaz. İlk tedaviden yıllar sonra ortaya çıkabilecek lokal nüks veya metastaz riski nedeniyle uzun süreli klinik ve radyolojik izlem gereklidir.
Prognoz
Genel olarak, AD'ler iyi bir prognoza sahiptir. Olguların yaklaşık %70'inde tam remisyon sağlanabilir. Bununla birlikte, vakaların %10-50'sinde, hastalık metastazlar nedeniyle ölümcül olabilir (karaciğerde, kemikte ve beyinde metastaz bildirilmiş olmasına rağmen en sık akciğerlerde ve lenf düğümlerinde görülür). Metastatik hastalığı olan hastaların ortalama sağkalımının 12 yıl olduğu bildirilmektedir.
Uzman(lar): Pr M. [Mario] MAAS - Pr R.R. [Rick] VAN RIJN – Son Güncelleme: Temmuz 2014
Çeviren: Ezgi Gizem BERKAY, MD – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalığın tanımı
Addison hastalığı (AH) adrenal korteksin otoimmün yıkımı yüzünden glukokortikoid ve mineralokortikoid eksikliği ile sonuçlanan kronik ve nadir hormonal rahatsızlıktır. Aslında AH otoimmün adrenaliti işaret eder fakat bu terim yaygın olarak kronik primer adrenal yetmezliğin (CPAI; bu terime bakınız) herhangi bir formunu tarifte kullanılır.
ORPHA:85138
- Hastalığın diğer adı/adları:
- Otoimmün Addison hastalığı
- Otoimmün adrenalit
- Klasik Addison hastalığı
- Primer Addison hastalığı
- Görülme Sıklığı: 1-5 / 10 000
- Kalıtımı: -
- Görülmeye başladığı yaşlar: Tüm yaşlar
- ICD-10: E27.1
- OMIM: 103230 240200
- UMLS: C00 01403 C0271737
- MeSH: D000224
- GARD: 5740
- MedDRA: 10001130
Özet
Epidemiyoloji
Addison hastalığının prevalansı gelişmiş ülkelerde 1/9,000-1/6,900’dir.
Klinik Açıklaması
Hastalığın başlama üst sınırı 40’lı yaşlardır fakat her yaşta da görülebilir. Spesifik olmayan semptomlarla sinsice meydana gelir ki diğer daha yaygın vakalarla karıştırılabilir. Yaygın belirtileri bitkinlik, enerji düşüklüğü, keyifsizlik, kilo kaybı, mide bulantısı, iştahsızlık (büyüyen çocuklarda), kas ve eklem ağrısını kapsar. Derinin ve muköz membranların pigmentasyonu (derinin özellikle avuç içi çizgilerinde, bilek eklemlerinde, yara izlerinde, oral mukoza ve sürtünme yerlerinde kararma) AH’nın esas belirtileridir. Postural hipotansiyon ve hipoglisemi semptomları hastalığın geç belirtileridir. Hastalar ayrıca tuzu çok tüketmek isteyebilir. Vitiligo ve alopesi areata sıklıkla mevcuttur. AH ayrıca sadece kadınlarda görülen ek semptomlara sebebiyet veren (aksiller/pubik bölgede kıl kaybı, çocuklarda puberş yokluğu, azalan libido ve kuru cilt) dehidroepiandrosteron eksikliğine neden olur. Akut primer adrenal yetmezlik (AAI; bu terime bakınız), adrenal kriz olarak da anılır, tedavi takip edilmediğinde ya da hastalığın süratlenme sürecinde hayati tehlike arz eden medikal bir acil durum ortaya çıkabilir.
Etiyoloji
AH adrenal korteksin otoimmün destrüksiyonundan kaynaklanır ve otoimmün bir bozukluğun parçası olarak görülebilir ya da ayrılabilir (otoimmün poliendokrin sendromu tip 1, 2 veya 4; bu terimlere bakınız).
Tanı yöntemleri
AH’nin tanısı için biyokimyasal testler elzemdir. Sabahın erken serum kortizolü ve plazma adenokortikotropik hormon (ACTH) düzeyleri ölçülür. AH olan kişilerde plazma ACTH seviyesi daha yüksektir (>22 pmol/L) ve sabah serum kortizol seviyeleri genelde düşüktür (
Ayırıcı tanı
Sekonder adrenal yetmezliğin elimine edilmesi gerekir. Sebepler hipofiz bezi tümörleri, lenfatik hipofizit, hipofiz tüberkülozu ve sarkoidozu içerir, bunların tümü ayırıcı tanıdır. İnfiltratif rahatsızlıklar ve CPAI’nın diğer sebepleri hariç tutulmalı ve tüberküloz (bu terime bakınız), mantar enfeksiyonları ve AIDS-ilişkili oportünist enfeksiyonlar dahil edilmelidir. Genetik bozukluklar, tümörler ve bazı ilaçlarla tedavi, CPAI’nın diğer daha az yaygın sebeplerindendir.
Kontrol ve tedavi
Kontrol ömür boyudur ve bir multidisipliner takım gerektirir. Oral hidrokortizonlu glukokortikoid replasmanı (günlük alım 10-25 mg ve 2-3 doz halinde) fizyolojik kortizol sekresyon paternlerini taklit etmek için verilir. Oral fludrokortizon, mineralokortikoid hormonlarının yerine kullanılır. Dehidroepiandrosteon isteğe bağlı yerine konur. Glukokortikoid seviyeleri AAI’yı önlemek için stres süresince ayarlanabilir. Hidrokortizonun dozu, hastanın refahı aşırı replasman veya az replasman belirtilerinin varlığı göz önünde bulundurularak klinik değerlendirme ve yanıtlar temelinde korunur. Plazma renin aktivitesinin değerlendirilmesi fludrokortizonun dozunun optimize edilmesinde yardımcıdır. Çocuklarda büyüme ve gelişme gözlenmelidir. Adrenal kriz ihtimaline karşı hastalar, enjeksiyona hazır bir hidrokortizon preparasyonu ve medikal bir uyarı kartı taşımalıdırlar.
Prognoz
AH’de iyileşme görülmez ancak AAI'yı önlemek için uygun tedavi ve özen gösterildiğinde yaşam süresi azalmaz. AH sadece görmezden gelindiğinde hayatı tehdit eder. Uzman hakem(ler): Dr Anne BACHELOT – Son güncelleme: Ekim 2012
Çeviren: Ayşe Nur Kurtcuoğlu – OCAK 2019
Kaynak sayfa için tıklayınız
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
Adrenal korteksten köken alan nadir bir kanserdir.
ORPHA: 1501
- Eş Anlamlısı(ları): -
- Prevalans: 1-9 / 100.000
- Kalıtım: Uygulanamaz
- Başlangıç yaşı: Çocukluk
- ICD-10: C74.0
- OMIM: 202300
- UMLS: C0206686
- MeSH: D018268
- GARD: 558
- MedDRA: 10001388
Özet
Epidemiyoloji
Çocukluk malinitelerinin %0,2’sine tekabül eder ve uluslararası insidansı 1/2.000.000’dur. Kızlarda erkeklerden daha sıktır. (oranı 1,5:1)
Klinik Tanım
Adrenokortikal karsinom (AKK), çocuklukta görülen ve 3,5 yaşında en üst insidans ile ortaya çıkar. Çocuklar ve ergenlerde çoğu AKK hormon salgılar ve klinik prezentasyonu tümör tarafından salgılanan hormon çeşidini yansıtır. Virilizasyon belirti ve semptomları vakalarda %90’ın üstünde mevcut olduğu gibi hipertansiyon da mevcut olabilir. Hirşutizm, akne ve seste kalınlaşma her iki cinsiyette de görülür. Kızlarda ayrıca kliteromegali ve yüzde kıllanma görülebilirken erkeklerde fallomegali ve erken virilizasyon mevcuttur. Cushing sendromu (bu terim için bakınız), vakaların üçte birinde, ay benzeri bir yüz görünümü, merkezcil yağ dağılımı ve bolluğu olan en yaygın belirtilerle ortaya çıkar. İzole hemihipertrofi, Beckwith-Wiedemann sendromu, Konjenital Adrenal Hiperplazi (KAH) ve Li-Fraumeni sendromu (LFS) olan hastalarda AKK insidansı artmıştır (bu terimlere bakınız). Tanısal Yöntemler Tanı, idrar ve kan testleri, deksametazon supresyon testi, abdomen ultrason, BT ve MR sonuçlarına dayanır.
Ayırıcı Tanı
Ayırıcı tanısında, feokromositoma ve nöroblastom (bu terimlere bakınız) vardır.
Genetik Danışmanlık
AKK’lerin en az %50’si kalıtsaldır ve bu nedenle bir kanser genetiği birimine sevk edilmesi önerilir.
Yönetim ve Tedavi
Cerrahi tam radikal rezeksiyon önerilen tedavidir ve özellikle küçük tümörler için iyileştirici olabilir. Eksik rezeksiyonlu ve metastatik yayılmalı vakalarda kemoterapi ve/veya mitotan (semptomatik ve ileri-rezekte edilemeyen, metastatik veya nüks- AKK’ler için yetim ilaç olarak AB pazarlama iznini 2004’de alan) tedavi seçenekleri arasındadır.
Prognoz
AKK, tamamen rezeke edilen küçük tümörler dışında kötü bir prognoza sahiptir (5 yıllık sağkalım %36'dır).
Uzman: Dr Bernadette BRENNAN – Son Güncelleme: Haziran 2007
Çeviren: Can Veysel ŞOROĞLU, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
ORPHA: 183669
- Eş anlamlısı(ları): -
- Prevalans: 1-9 / 1.000.000
- Kalıtım: -
- Başlangıç Yaşı: -
- ICD-10: -
- OMIM: -
- UMLS: C0001768
- MeSH: D000361
- GARD: -
- MedDRA: 10001471
Özet
Şu anda bu hastalık için bir Orphanet özeti geliştirilmektedir. Hastalığa ilişkin diğer verilere linkteki sayfada yer alan Ek bilgi menüsünden ulaşılabilir.
Çeviren: Can Veysel ŞOROĞLU, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
Farklı dokularda mast hücrelerinin önemli miktarda infiltrasyonu ile karakterize edilen nadir ve ağır bir sistemik mastositoz (SM) formu.
ORPHA: 98850
- Eş Anlamlısı(ları): -
- Prevalans: 1-9 / 1.000.000
- Kalıtım: Uygulanamaz
- Başlangıç Yaşı: Yetişkin
- ICD-10: C96.2
- OMIM: -
- UMLS: C1112486
- MeSH: -
- GARD: -
- MedDRA: 10056453
ÖZET
Epidemiyoloji
SM vakalarının %10’dan azına tekabül eder ve küresel prevalansı 1/400.000 ve 1/250.000 arasında tahmin edilmektedir.
Klinik Tanım
Agresif sistemik mastositoz (ASM) genellikle çocuklarda gelişmez. Kutanöz tutulum normalde yoktur. Hastalarda mast hücre invazyonu ve yoğun medyatör salınımı ile senkop, tekrarlayan kızarıklık ishal, ağrı, organomegali ve organ fonksiyon bozukluğu, hematopoetik fonksiyon bozukluğu (izole sitopeniden belirgin pansitopeniye kadar farklılık gösteren kan sayımının bozulmasına neden olabilir), kırık riski osteoporoz biçiminde kemik tutulumu ve malabsorpsiyon görülür. ASM’nin en ciddi komplikasyonu büyük ihtimalle ölümcül anafilaktik şoktur. Hastalar mast olmayan hücreli hematolojik hastalık belirtileri göstermezler.
Etiyoloji
ASM etiyolojisi iyi anlaşılamamıştır ancak ASM’li hastaların büyük çoğunluğunun mast hücrelerinde aktive edici KIT mutasyonu, genellikle D816V, olduğuna dair kanıtlar vardır.
Tanısal Yöntemler
Tanı, kemik iliğinin histolojik ve sitolojik analizine dayanır. Sitolojik analizle kemik iliği mast hücrelerinin %5’i ila 20’si açıklanır ve bu çoğu ASM vakasında, kemik iliği mast hücreleri tek veya çok loblu bir çekirdek veya bazen blast benzeri bir morfolojik görünümde atipik olgunlaşmamış görünüme sahiptir. Serum triptaz ölçümü sürekli 20ng/ml’nin üzerindedir ve aktive edici KIT mutasyonu vakaların büyük çoğunluğunda pozitiftir.
Ayırıcı Tanı
Ayırıcı tanısı, tüm sitopeni nedenlerinin (miyelofibroz, miyelodisplazi ve diğer hematolojik maliniteler; bu terimlere bakınız) ve JAK2’deki diğer patojenik mutasyon tiplerini içerir. Aktive edici KIT mutasyonu ile kemik iliğinin mast hücrelerince infiltrasyonunun saptanmasıyla dışlanır.
Yönetim ve Tedavi
Kısmen semptomatik tedavi, ağırlıklı olarak antihistaminikler (anti-H1 ve Anti-H2) ve kısmen antiproliferatif (cladribine kullanılan kortikosteroidli veya kortikosteroidsiz interferon alfa tedavisi) tedavi uygulanır. KIT D816V mutasyonu görülmeyen birkaç vaka, imatinib mesilat veya masitinib mutasyona uğramamış KIT formunu inhibe ettiği için kullanılır. Mutant D816V formunu inhibe eden moleküller klinik çalışmalarla incelenmektedir ve diğer terapötik yaklaşımlar (rapamisin, bortezomib veya talidomid kullanımı dahil) önerilmiştir. İyotlu kontrast ajanların kullanımı sınırlı olmalıdır, çünkü mast hücre medyatörlerinin salınımını ciddi arttırır.
Prognoz
İki ila dört yıllık ortalama sağkalım ile kötü prognozludur.
Uzman: Pr Michel AROCK – Son güncelleme: Kasım 2008
Çeviren: Can Veysel ŞOROĞLU, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
Bazal gangliyon kalsifikasyonu, lökodistrofi ve beyin omurilik sıvısı (BOS) lenfositozu ile karakterize kalıtsal, subakut ensefalopati.
ORPHA: 51
- Eş anlamlıları:
- Bazal gangliyon kalsifikasyonlu ensefalopati
- İntrakraniyal kalsifikasyon ve beyin omurilik sıvısının kronik lenfositozlu ensefalopati
- Prevalans: 1-5 / 10.000
- Kalıtım: Otozomal dominant veya Otozomal resesif
- Başlangıç yaşı: İnfant, Yeni Doğan
- ICD-10: G31.8
- OMIM: 114100 225750 610181 610329 610333 612952 615010 615846
- UMLS: C0393591
- MeSH: C535607
- GARD: 575
- MedDRA: -
Özet
Epidemiyoloji
Şimdiye kadar literatürde 120’den fazla vaka bildirilmiştir.
Klinik Tanım
Etkilenen bebeklerin çoğunluğu normal büyüme parametreleri ile zamanında doğarlar. Hastalık başlangıcı epilepsi (vakaların %53), ekstremitelerde “chilblain” deri lezyonları (vakaların %43) ve aseptik ateşli hastalık atakları (vakaların %40) ile ilişkili şiddetli, subakut ensefalopati (beslenme sorunları, sinirlilik ve psikomotor regresyon veya gecikme) ile yaşamın ilk birkaç gün veya ay içinde ortaya çıkar. Semptomlar, hastalık seyri stabilize edilmeden önce birkaç ay içinde (mikrosefali ve piramidal bulguların gelişmesiyle birlikte) ilerlemektedir. Bununla birlikte, 1 yaş sonrası başlangıçlı, dil becerilerinin ve bilişsel işlevin korunduğu, normal baş çevresi ile seyreden daha az şiddetli formlar tanımlanmıştır. Fenotip, aile içi ve aileler arası varyasyon gösterir.
Etiyoloji
2006’da dört gende hastalığa neden olan mutasyonlar tanımlandı: 3'->5' ekzonükleaz kodlayan TREX1 ve Rnaz H2 endonükleaz kompleks alt birimlerini kodlayan genler olan RNASEH2A, RNASEH2B ve RNASEH2C. TREX1 (vakaların %25'i), RNASEH2C (vakaların %14’ü) ve RNASEH2A (vakaların %4’ü) mutasyonları ciddi bir fenotipe neden olurken, RNASEH2B (vakaların %41’i) mutasyonları genellikle daha hafif bir fenotipe yol açar. Kalan vakalarda bu genler için mutasyon bulunmaz.
Tanı Yöntemleri
Kalsifikasyon (bazal gangliyon ve beyaz maddeyi içeren), kistik lökodistrofi (ağırlıklı olarak frontotemporal) ve kortikal-subkortikal atrofi tanı için ana bulgulardır ve genellikle korpus kallosum, beyin sapı ve serebellumun atrofisi ile ilişkilidir.
Ayırıcı Tanı
Temel ayırıcı tanısı TORCH konjential enfeksiyonlarını (toksoplazma, rubella, CMV, HSV1 ve HSV2) kapsar.
Antenatal Tanı
Prenatal tanısı amniyotik sıvı (amniyosentez) veya trofoblastların (koryon villus aspirasyonu) moleküler analizi ile mümkündür.
Genetik Danışma
Otozomal resesif olarak aktarılsa da nadiren otozomal dominant kalıtım vakaları da bildirilmiştir.
Yönetim ve Tedavi
Tedavi semptomatiktir (beslenme problemlerinin yönetimi, psikomotor gecikme ve varsa epilepsi).
Prognoz
Şiddetli formla başvuran hastaların yaklaşık %80'i yaşamın ilk 10 yılında ölmektedir, ancak yaşamın ilk on yılından sonra uzun süreli sağkalım daha hafif formlarda bildirilmiştir.
Uzmanlar: Dr Pierre BLANC - Pr Isabelle CREVEAUX – Son güncelleme: Haziran 2008
Çeviren: Can Veysel ŞOROĞLU, MSc – OCAK 2019
Kaynak sayfa için tıklayınız
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
ORPHA:90081
- Eş anlamlısı: -
- Prevalans: 1-5 / 10.000
- Kalıtım: -
- Başlangıç yaşı: -
- ICD-10: B22.2
- OMIM: -
- UMLS: C0343755
- MeSH: -
- GARD: -
- MedDRA: -
Özet
Bu, Avrupalı ve/veya Amerikalılarda yetim (orphan) olarak adlandırılan nadir rastlanan bir durumdur.
Çeviren: Can Veysel ŞOROĞLU, MSc – OCAK 2019
Kaynak sayfa için tıklayınız
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
Ailesel adenomatoz polipozis (FAP/AAP), yaşamın ikinci on yılında rektum ve kolonda yüzlerce ila binlerce adenom gelişimi ile karakterizedir.
ORPHA: 733
Eş Anlamlısı(ları):
Kolorektal adenomatoz polipozis
FAP
Ailesel polipozis koli
Prevalans: 1-9 / 100.000
Kalıtım: Otozomal dominant ya da otozomal resesif
Başlangıç yaşı: Yetişkin
ICD-10: D12.6
OMIM: 175100
UMLS: C0032580
MeSH: D011125
GARD: 6408
MedDRA: 10056981
Özet
Epidemiyoloji
FAP yaklaşık 1/8.300 doğum insidansına sahiptir, her iki cinsiyette de aynı şekilde görülür ve kolorektal kanser (KRK) vakalarının %1'inden azını oluşturur. AB'de yaygınlık 1/11.300-1/37.600 olarak tahmin edilmektedir.
Klinik Tanım
Hastaların çoğu, adenomlar büyük ve sayısız olana kadar yıllarca asemptomatiktir ve rektal kanamaya, hatta anemiye veya kanser gelişimine neden olur. Genellikle, kanser poliplerin ortaya çıkmasından on yıl sonra gelişmeye başlar. Spesifik olmayan semptomlar kabızlık veya ishal, karın ağrısı, elle hissedilen karın kitleleri ve kilo kaybını içerebilir. FAP osteomalar, dental anormallikler, retinal pigment epitelinin konjenital hipertrofisi (RPEKH), desmoid tümörler ve ekstrakolonik kanserler (tiroid, karaciğer, safra kanalları ve merkezi sinir sistemi) gibi ekstraintestinal belirtiler gösterebilir. Daha az agresif bir FAP varyantı, zayıflatılmış FAP (AFAP; bu terime bakınız), daha az kolorektal adenomatöz polipleri (genellikle 10 ila 100), daha sonra adenom görünüm yaşı ve düşük kanser riski ile karakterize edilir. Bazı lezyonlar (kafa derisi ve mandibula osteomları, diş anormallikleri, kafa derisindeki fibromlar, omuzlar, kollar ve sırt) Gardner sendromunun göstergesidir, oysa FAP ve medulloblastom arasındaki ilişki Turcot sendromu olarak adlandırılır (bu terimlere bakınız).
Etiyoloji
Klasik FAP, APC genindeki (5q21-q22) bir germ hattı mutasyonundan kaynaklanır. Hastaların çoğunda (yaklaşık %70) ailesinde kolorektal polip ve kanser öyküsü vardır. Bir hasta alt kümesinde, bir MUTYH mutasyonu (1p34.1), her ikisinde de az miktarda artmış CRC ve polip/adenom gelişme riski ile karakterize olan, resesif olarak kalıtsal bir polipoz durumuna, MUTYH ile ilişkili ailesel üst ve alt gastrointestinal sistem adenomatoz polipozuna (bu terime bakınız) neden olur.
Tanısal Yöntemler
Teşhis aile öyküsüne, klinik bulgulara ve kalın bağırsak endoskopisine veya tam kolonoskopiye dayanmaktadır. Mümkün olduğunda, klinik tanı genetik testlerle doğrulanmalıdır.
Ayırıcı Tanı
Ayırıcı tanılar çoklu poliplere neden olan diğer hastalıkları içerir (Peutz-Jeghers sendromu, ailesel çocuk polipozu veya hiperplastik polipoz, kalıtsal karışık polipoz sendromu ve Lynch sendromu; bu terimlere bakınız).
Antenatal Tanı
Presemptomatik ve prenatal (amniyosentez ve koryon villüs örneklemesi) ve hatta preimplantasyon genetik testleri mümkündür. Bir genetik uzmanına veya genetik danışmana başvurmak zorunludur.
Genetik Danışmanlık
Klasik FAP, otozomal dominant bir şekilde kalıtsaldır. Ailede APC mutasyonu tespit edildiğinde, tüm birinci derece akrabaların genetik testleri yapılmalıdır.
Yönetim ve Tedavi
Kanser önleme ve iyi bir yaşam kalitesini sürdürme, yönetimin ana hedefleridir ve tüm hastalara düzenli ve sistematik takip ve destekleyici bakım önerilmelidir. Gençlerin sonlarında veya yirmili yaşların başında, kolorektal kanser profilaktik ameliyatı savunulur. Önerilen alternatifler total proktokolektomi ve AFAP için ileoanal kese veya ileorektal anastomozdur. Duodenal kanser ve desmoidler total kolektomi sonrası iki ana ölüm nedenidir. Erken teşhis edilmeleri ve tedavi edilmeleri gerekir. Ampullar ve duodenal kanser riskini azaltmak için üst endoskopi gereklidir. Progresif tümörleri ve geri dönüşümsüz hastalığı olan hastalar, sitotoksik kemoterapi ve cerrahi kombinasyonuyla cevap verebilir veya stabilize olabilir. Celecoxib, ABD Gıda ve İlaç İdaresi (FDA) ve Avrupa İlaç Ajansı tarafından FAP hastalarında ek tedavi olarak kullanılmak üzere pazarlama izni aldı.
Prognoz
FAP'lı hastalar % 100 KRK riski taşırlar. Ancak, hastalar bir tarama tedavi programına girdiklerinde bu risk önemli ölçüde azalır.
Uzman(lar): Dr Dani BERCOVICH - Dr Elizabeth HALF - Pr Paul ROZEN–Son Güncelleme: Ekim 2009
Çeviren: Ezgi Gizem BERKAY, MD
ORPHA: 93372
Eşanlamlısı(ları) :AHH tip 1
Prevalans: 1-9 / 100.000
Kalıtım: Otozomal dominant
BaşlangıçYaşı: Tüm yaşlar
ICD-10: E83.5
OMIM: 145980
UMLS: C0342637 C1809471
MeSH: C537145
GARD: 2796
MedDRA: 10068704
Özet
Bu hastalık ailesel hipokalsiürik hiperkalsemi altında tanımlanmaktadır.
Çeviren: Ezgi Gizem BERKAY, MD
Hastalık Tanımı
Ailesel izole dilate kardiyomiyopati, sol ve/veya sağ ventriküllerin sistolik ve diyastolik disfonksiyonuna yol açan, kalp yetmezliği veya aritmiye neden olan dilatasyon ile karakterize, nadir görülen, genetik olarak heterojen bir kalp hastalığıdır.
ORPHA: 154
Eş anlamlısı(ları) : Ailesel veya idiyopatik dilate kardiyomiyopati
Prevalans:1-5 / 10.000
Kalıtım: Otozomal dominant / Otozomal resesif / X’e bağlı resesif / Mitokondriyal kalıtım
Başlangıç Yaşı: Tüm yaşlar
ICD-10: I42.0
OMIM: 115200 302045 600884 601154 601493 601494 604145 604288 604765 605582 606685 607482 608569 609909 609915 611407 611615 611878 611879 611880 612158 612877 613122 613172 613252 613286 613424 613426 613642 613694 613697 613881 614672 615184 615235 615248 615373 615396 615916 618189
UMLS: C0340427
MeSH: -
GARD: 2905
MedDRA: -
Çeviren: Ezgi Gizem BERKAY, MD
ORPHA: 75249
Eşanlamlısı(ları) :Ailesel veya idiyopatik kısıtlayıcı kardiyomiyopati
Prevalans:1-9 / 100.000
Kalıtım: Otozomal dominant veya uygulanamaz
BaşlangıçYaşı: Tümyaşlar
ICD-10: I42.5
OMIM: 115210 609578 612422 615248 617047
UMLS: -
MeSH: -
GARD: -
MedDRA: -
Özet
Bu hastalık için bir Orphanet özeti halen geliştirilme aşamasındadır. Bununla birlikte, hastalıkla ilgili diğer verilere bu sayfanınaltındaki Ek Bilgi menüsünden erişilebilir.
Çeviren: Ezgi Gizem BERKAY, MD
Hastalık Tanımı
Ailesel Kısmi Lipodistrofi (AKLD), çoğu durumda, ekstremitelerden ve kalçalardan, çocukluktan veya erken yetişkinlikten yağ kaybıyla karakterize edilen ve genellikle akantoz nigrikans, insülin direnci, diyabet, hipertrigliseridemi ve karaciğer steatozu ile ilişkili nadir genetik lipodistrofik sendrom grubudur.
ORPHA:98306
Eş Anlamlısı(ları):AKLD
Prevalans: 1-9/100.000
Kalıtım: Otozomal dominant ya da otozomal resesif
Başlangıç yaşı: -
ICD-10: E88.1
OMIM: -
UMLS: C0271694
MeSH: D052496
GARD: 11962
MedDRA: -
Özet
Epidemiyoloji
AKLD prevalansı bilinmemektedir, ancak Dunnigan tipi AKLD (AKLD2; bu terime bakınız) en yaygın formdur. Kobberling tipi AKLD (AKLD1; bu terime bakınız) açıkça tanımlanmamıştır ve farklı etiyolojik alt tipleri içerebilir. Bir PPARG mutasyonu kaynaklı AKLD (AKLD3) yaklaşık yirmi ailede tanımlanmıştır. PLIN1 mutasyonları nedenli AKLD (AKLD4) bugüne kadar 4 ailede tanımlanmıştır. CIDEC'e bağlı AKLD mutasyonları (AKLD5) bir hastada ve iki ailede LIPE mutasyonları (AKLD6) nedeniyle FPLD tanımlanmıştır. İnsüline dirençli diyabet ve parsiyel lipodistrofi olan bir ailede AKT2 heterozigot mutasyonu bildirilmiştir.
Klinik Tanım
AKLD, klinik ve genetik olarak heterojen bir nadir bozukluk grubudur. En yaygın ve iyi tarif edilen formu (AKLD2), ergenlikte veya ergenliğe yakın dönemde lipodistrofi başlangıcını, uzuvlardan, kalçalardan ve gövdeden subkutanöz yağ dokusunun bölgesel kaybını ve devamında yüz, boyun ve aksiller bölgelerde ilerleyici bir yağ birikimini içerir. Hastalara cushingoid bir görünüm verir. Artmış iskelet kası hacmi ve kütlesi de gözlenmiştir. Ekstremitelerde belirgin damarlar (lipoatrofi nedeniyle) görülür. Metabolik komplikasyonlar ergenlik döneminde veya yetişkinlikte kademeli olarak ortaya çıkar ve insülin direnci, diyabet, hepatik steatoz, akantoz nigrikans, yüksek tansiyon ve koroner kalp hastalığı riski artmış erken ateroskleroz içerir. Bazı kadınlar, hirşutizm, oligomenore, polikistik overler ve infertilite gibi polikistik over sendromunun özelliklerini gösterebilir. Hastalar tipik olarak erken kardiyovasküler hastalıklara yatkındır. Diğer belirtileri diyabet, tekrarlayan akut pankreatit ve karaciğer steatohepatit veya siroz komplikasyonları içerebilir. Diğer AKLD formları benzer metabolik komplikasyonlarla ilişkilidir, ancak lipoatrofi daha sık normal veya artmış truncal yağ ile ekstremitelerle sınırlıdır. AKLD3 sıklıkla şiddetli hipertansiyon ile ilişkilidir.
Etiyoloji
AKLD'nin en yaygın formu olan AKLD2, hücresel ara filamentler A tipi laminleri kodlayan LMNA genindeki (1q22) bir mutasyondan kaynaklanır. Bu gendeki diğer mutasyonlar, otozomal kodominant şiddetli lipodistrofik laminopati gibi diğer laminopatik fenotiplerle (kas ve/veya kardiyak distrofiler, hızlanmış yaşlanma) daha sık ilişkili atipik lipodistrofilere yol açabilir. AKLD'nin daha nadir genetik varyantları ile ilişkili diğer genler arasında PPARG (3p25) (AKLD3), AKT2 (19q13.1-q13.2), PLIN1 (15q26) (AKLD4), CIDEC (3P25) (AKLD5) ve LIPE (19q13.1-q13.2) (AKLD6) bulunur. AKLD1 etiyolojisi bilinmemektedir. AKLD fenotipli birçok hastada bu genlerde mutasyon yoktur ve bu nedenle yeni AKLB genleri henüz keşfedilmemiştir.
Genetik Danışmanlık
İlgili mutasyona bağlı olarak, AKLD otozomal dominant veya resesif bir şekilde kalıtılabilir. Bilinen bir mutasyona sahip ailelerde genetik danışmanlık mümkündür.
Uzman(lar): Pr Corinne VIGOUROUX–Son Güncelleme: Şubat 2015 Çeviren: Ezgi Gizem BERKAY, MD
Hastalık Tanımı
Ailesel melanom (FM), etkilenen bir ailede iki birinci derece akrabada veya daha fazla akrabada histolojik olarak doğrulanmış melanom gelişimi ile karakterize nadir bir kalıtsal melanom şeklidir.
ORPHA: 618
Eş Anlamlısı(ları):-
Prevalans: Bilinmiyor
Kalıtım: Otozomal dominant ya da multigenik/multifaktöryel
Başlangıç yaşı: Yetişkinlik
ICD-10: C43.0 C43.1 C43.2 C43.3 C43.4 C43.5 C43.6 C43.7 C43.8
OMIM: 155600 155601 155700 608035 609048 613099 613972 615134 615848
UMLS: C2314896
MeSH: -
GARD: -
MedDRA: -
Özet
Epidemiyoloji
FM'nin tüm kutanöz melanom vakalarının yaklaşık %10'unu oluşturduğu düşünülmektedir. Melanom en çok Avrupa kökenli popülasyonları etkiler. Daha fazla güneşe maruz kalınan (ABD’nin güneyi, Avustralya, Yeni Zelanda) coğrafi bölgelerde insidansı daha yüksektir. İnsidans 2012 yılında Avrupa'da 1/90.000 olarak tahmin edilmiştir.
Klinik Tanım
Ailesel melanom, ailesel olmayan melanomdan daha erken ortaya çıkma eğilimindedir. Ortalama başlangıç yaşı genellikle 30 ile 40 yıl arasında iken, ailesel olmayan melanom tipik genel popülasyonda 50 ile 60 arasında görülür. Bazı ailelerde daha erken ve daha geç başlangıçlı vakalar bildirilmiştir. Çoklu primer melanomların daha yüksek frekansı olduğu da bulunmuştur. Melanom genellikle ciltte pigmentli bir lezyon olarak ortaya çıkar. Genellikle düzensiz sınırlar ve renk çeşitliliği ile asimetriktir. Çap genellikle 6 mm'den büyüktür. En yaygın alanlar gövde, alt bacaklar ve sırttır. Tanımlanan primer kutanöz melanomun (yayılan melanom, nodüler melanom, lentigo malign melanom ve akral lentiginöz melanom) dört ana klinikopatolojik alt tipinin tümü gözlenir. Çeşitli büyüme kalıpları ile Diğer organlara metastaz potansiyeline ilerleme görülür. Atipik nevüsler ailesel melanomlu ailelerde sıkça bulunur.
Etiyoloji
Ailesel melanom riski, yatkınlık genlerinde çok çeşitli genetik değişikliklerle yakından ilişkilidir, aynı zamanda pigmentasyon, çiller, nevüsler ve güneş reaksiyonları gibi fenotipik risk faktörlerinden etkilenmiş gibi görünmektedir, ancak UV radyasyonuna da maruz kalmaktadır. FM'nin altında bu nedenle genetik ve çevresel faktörler arasındaki karmaşık etkileşimlerin yattığı düşünülmektedir. FM'de yer alan en yaygın yüksek penetranslı duyarlılık geni, FM'nin yaklaşık %20'sinde yatkınlığı oluşturan CDKN2A'dır. CDK4, başka bir yüksek riskli gen, nadiren yer almaktadır. BAP1, POT 1, TERF2IP, ACD ve TERT mutasyonları yakın zamanda bildirilmiştir ve penetransı belirlenmeye devam etmektedir. MC1R orta penetranslı genlerdendir. Bazı yaygın yirmi genetik varyant düşük kümelenmiş ailelerde melanom riskini düzenler.
Tanısal Yöntemler
İki veya daha fazla yakın akrabasında melanom gelişen bireylerde ailesel melanomdan şüphelenilir. Melanomu tanımlamak için yeni veya değişen bir cilt yapısı değerlendirilmelidir. Değişiklikler renk, sınır, boyut ve simetri içerir.
AyırıcıTanı
Ana ayırıcı tanılar seboreik keratoz, atipik leke, ailesel atipik çoklu leke melanomu ve cilt karsinomudur.
Genetik Danışmanlık
Etkilenen bazı ailelerde melanom kalıtımı otozomal dominanttır ancak çoğu durumda poligenik bir kalıtım modeli olasıdır. Yakın akrabalarında melanom öyküsü olan hastalara ortalama 2 kat artmış riske sahip oldukları bildirilmelidir. Çoklu pigmentli atipik melanositik lezyonlar gibi birden fazla göreceli ve ilgili kişisel fenotipik öykü, önemli ölçüde daha yüksek bir riskle sonuçlanır.
Yönetim ve Tedavi
FM ailelerinde yüksek riskli bireylerin uzun süreli taranması ve izlenmesi önerilir. Altı ayda bir nitelikli bir dermatolog tarafından tüm vücut cilt muayenesi gereklidir. Cildin kişi tarafından muayenesi teşvik edilmelidir. Tedavi sporadik melanom tedavisine benzerdir ve büyük ölçüde başlangıç evrelerinde ameliyata dayanmaktadır.
Prognoz
Prognoz değişkendir; tanı, kalınlık, ülserasyon varlığı, mitotik indeks, olası lenf nodu tutulumu ve uzak metastatik hastalık varlığına bağlıdır. Erken tanı (ince Breslow kalınlığında) yüksek oranda tedavi ile ilişkilidir. Melanomlu ailelerin izlenmesi, olumlu bir prognoz ile sonuçlanan cilt takibi melanomu tespit etmeyi amaçlamaktadır.
Uzman(lar): Pr Marie-Françoise AVRIL - Pr Eve MAUBEC–Son Güncelleme: Şubat 2016
Çeviren: Ezgi Gizem BERKAY, MD
Hastalık Tanımı
Asemptomatik olabilen veya nöbetler, spesifik olmayan baş ağrıları, progresif veya geçici fokal nörolojik defisitler ve/veya beyin kanaması gibi değişken nörolojik belirtilere neden olabilen kümelenmiş düzensiz dilate kapilerler ile karakterize nadir, kılcal-venöz malformasyonlardır.
ORPHA:221061
Eş Anlamlısı(ları):
Ailesel beyin kavernöz anjiyomu
Ailesel serebral kavernom
Kalıtsal beyin kavernöz anjiyom
Kalıtsal serebral kavernom
Kalıtsal serebral kavernöz malformasyon
Prevalans: 1-5 / 10.000
Kalıtım: Otozomal dominant
Başlangıç yaşı: Tüm yaşlar
ICD-10: Q28.
OMIM: 116860 603284 603285
UMLS: C2931263
MeSH: -
GARD: -
MedDRA: -
Özet
Epidemiyoloji
Tüm serebral kavernöz malformasyonların (CCM/SKM) genel prevalansının 1/200 ila 1/1.000 olduğu tahmin edilmektedir. Ailesel serebral kavernöz malformasyon (FCCM/ASKM), tahmin edilen 1/5.000-1/10.000 prevalansı olan tüm SKM vakalarının yaklaşık %20'sini temsil eder ve bu nedenle sporadik SKM'lerin aksine nadir görülür. Hispanik-Amerikan SKM ailelerinde güçlü bir kurucu etkisi bulunmuştur.
Klinik Tanım
FSKM hastalarının %60'a yakını semptomatiktir. FSKM genellikle 20-30 yaşları arasında tezahür eder, ancak herhangi bir yaşta klinik belirtiler görülebilir. Belirtiler nöbetleri (%40-70), spesifik olmayan baş ağrıları (%10-30), ilerleyici veya geçici fokal nörolojik defisitleri (%35-50) ve/veya beyin kanamasını (%41) içerir. FSKM hastaları genellikle birkaç milimetreden birkaç santimetreye kadar değişen çok sayıda lezyonla ortaya çıkarlar. FSKM'ler baskın olarak beyinde görülür ancak aynı zamanda omurilik, retina (%5'inde) ve ciltte de bildirilmiştir.
Etiyoloji
Bugüne kadar, üç gendeki mutasyonların ailesel SKM'ye neden olduğu gösterilmiştir; Sırasıyla 7q21.2, 7p13 ve 3q26.1 kromozomu üzerinde bulunan KRIT1, CCM2 ve PDCD10 genleridir, çeşitli fonksiyonlarının arasında vasküler endotel hücreleri arasındaki bağlantı oluşumunu modüle eden proteinleri kodlar.
Tanısal Yöntemler
SKM'leri ortaya çıkaran serebral manyetik rezonans görüntüleme (MRG), SKM'yi teşhis etmek için altın standart bir araştırmadır ve hemosiderin için oldukça hassas olan T2 gradyan eko dizisi içermelidir. MRG, çoğu ASKM hastasında, yalnızca bir lezyonu barındıran sporadik olguların aksine çoğul lezyonları göstermektedir. Bu nedenle çoklu SKM lezyonlarının tespiti, hastalığın genetik yapısını kuvvetli bir şekilde ortaya koymaktadır. ASKM genlerinin moleküler taranması bazen atipik MRG lezyonları gösteren hastalarda tanıyı belirlemek için yararlıdır; bununla birlikte, çoğu durumda, genetik danışma için kullanılır.
Ayırıcı Tanı
Atipik hemorajik MRG lezyonları ile başvuran olgularda ASKM'nin ayırıcı tanısında çoklu hemorajik metastazlar veya amiloidozlu kalıtsal serebral kanama vardır.
Antenatal Tanı
Prenatal tanı mümkündür. Bununla birlikte, pratikte, bu hastalıkta çok az doğum öncesi tanı istenmektedir (çoğunlukla birkaç hastanın bazal ganglionlardaki veya omurilikteki veya ponstaki SKM'lerden ciddi şekilde etkilendiği ailelerde).
Genetik Danışmanlık
ASKM, eksik penetrasyonu olan otozomal dominant bir özellik olarak aktarılır. Etkilenen ailelere, mutasyona uğramış genin kalıtım riskinin %50'sini bildiren genetik danışmanlık verilmelidir. SKM'lerin genetik yatkınlığının değerlendirilmesindeki diğer önemli hususlar, MRG beyin taramasında lezyon sayısını, SKM klinik özelliklerinin ailesinde öyküyü ve başlangıç yaşını içerir.
Yönetim ve Tedavi
Genelde yılda bir kez MRG ile düzenli kontrol, SKM'nin fark edilmesinden sonra zaman zaman ek asemptomatik lezyonlar görülebileceğinden önerilmektedir. Bu MRG kontrolleri, eş zamanlı semptomların yokluğunda her 5 yılda bir verilebilir. Nöbet ve baş ağrısı tedavisi semptomatiktir. Ciddi sakatlayıcı nöbetlere ve/veya fokal nörolojik defisitlere ve/veya beyin kanamalarına neden olan lezyonlar, mümkün olduğunda lezyonların cerrahi olarak çıkarılmasını gerektirir. Asetilsalisilik asit, heparin ve varfarin kullanımı kanama riskini artırabilir.
Prognoz
ASKM, hastanın yaşı ile SKM lezyonlarının sayısı arasında güçlü bir korelasyon ile gelişen bir durumdur. Hemorajik olay oranının, her yıl lezyon başına %2-5 olduğu tahmin edilmektedir. Fonksiyonel sonuç çoğunlukla SKM lezyonlarının yeri ile ilgilidir, beyin sapı ve bazal ganglion lezyonları kötü prognozludur. Mevcut veriler, çoğu hastada uzun vadeli prognozun, vakaların %80'inde korunmuş bir otonomi için oldukça uygun olduğunu göstermektedir.
Uzman(lar): Pr Elisabeth TOURNIER-LASSERVE–Son Güncelleme: Ağustos 2019
Çeviren: Ezgi Gizem BERKAY, MD
Hastalık Tanımı
Ailesel tiroit dishormonogenezi birincil konjenital hipotiroidizm türüdür (bu terime bakınız), doğumdan itibaren mevcut olan kalıcı bir tiroid hormonu eksikliği, tiroit hormon sentezinin doğuştan gelen hatalarından kaynaklanır.
ORPHA:95716
Eş Anlamlısı(ları):Tiroit dishormonogenez
Prevalans: 1-9 / 100.000
Kalıtım: Otozomal resesif
Başlangıç yaşı: Yenidoğan, bebeklik
ICD-10: E03.0 E03.1
OMIM: 274400 274500 274700 274800 274900 607200
UMLS: -
MeSH: -
GARD: -
MedDRA: -
Özet
Epidemiyoloji
Tiroit dishormonogenezi, kalıcı konjenital hipotiroidizmin %10-15'ini oluşturur (bu terime bakınız).
Klinik Tanım
Klinik bulgular, konjenital hipotiroidizmin diğer biçimleridir (bu terime bakınız). Hipotiroidizm özelliklerine ek olarak, hastalarda dishormonogenezle birlikte guatr da ortaya çıkabilir.
Etiyoloji
Dishormonogenez, çoğunlukla otozomal resesif kalıtılır, ancak en az bir durumun otozomal dominant kalıtımı olan tiroit hormon sentezi ve sekresyon basamaklarındaki kalıtsal kusurlardan kaynaklanabilir. En sık görülen kusur, toplam iyot organifikasyon defektlerine (TİOD) yol açan ve DUOX2 ve DUOX2A genlerinde (15q15.3, 15q15) otozomal dominant mutasyonlardan kaynaklanabilen tiroit peroksidaz aktivitesidir. Daha az ciddi kusurlar kısmi iyot organifikasyon kusurlarına (PİOK) neden olur ve sodyum/iyodür taşınmasında kusurlar, kusurlu tiroglobulin eylemi veya iyodotirozindeiodinaz enziminde bir kusur içerebilir.
Tanısal Yöntemler
Yenidoğan tarama programları olan ülkelerde (primer tiroksin (T4)-takip tiroit uyarıcı hormon (TSH) veya primer TSH testi ile), konjenital hipotiroidi olan bebekler tarama testleri ile tespit edildikten sonra teşhis edilir. Bununla birlikte, yenidoğan taramasıyla tespit edilen bebeklerde guatr nadiren görülür. Tanı, yüksek serum TSH seviyesi ve düşük T4 veya serbest T4 seviyesi ile doğrulanmalıdır. Tanı, tiroit bezinin yüksek radyoaktif iyot (RAI) alımına ve ardından sodyum perklorat uygulamasından sonra %90'dan fazla salınmaya dayanır. Genetik testler de kullanılabilir. PID, perklorat uygulamasından sonra %50-90 salınımı ile teşhis edilir (ve genetik testlerle doğrulanabilir).
Ayırıcı Tanı
Ayırıcı tanılar diğer konjenital hipotiroidizm biçimlerini içerir (bu terime bakınız)
Antenatal Tanı
Genetik bir kusurun tespit edildiği ailelerde genetik danışmanlık ve antenatal tanı sunulabilir.
Yönetim ve Tedavi
Levotiroksin, serum T4'ü 130 nmol/L'nin (10 ug/dL) üzerinde olabildiğince hızlı bir şekilde yükseltmek için acil bir hedefle (10-15mcg/kg/gün başlangıç dozu) tercih edilen tedavidir; bu dozlarda, serum TSH genellikle 2-4 hafta içinde normalleşir. Bebeklik döneminde sık laboratuvar izleme optimal nörobilişsel sonucu sağlamak için esastır. Serum TSH ve T4 veya serbest T4, yaşamın ilk 6 ayında her 1-2 ayda bir, 6 ay ile 3 yaş arasındaki her 3 ayda bir ve herhangi bir doz değişikliğinden 4 hafta sonra ölçülmelidir.
Prognoz
Erken tedaviye başlayan bebeklerin prognozu, kardeş veya sınıf arkadaşı kontrollerine benzer IQ'larla mükemmeldir. Daha sonraki bir yaşta (>30 gün) başlayan bebeklerde, şu anda önerilenden daha düşük L-tiroksin dozlarında ve daha şiddetli hipotiroidi olan bebeklerde daha düşük nörobilişsel sonuçlar ortaya çıkabilir.
Uzman(lar): DrStephen LAFRANCHI - DrMaynika RASTOGI –Son Güncelleme: Ağustos 2010
Çeviren: Ezgi Gizem BERKAY, MD
Hastalık Tanımı
Konjenital uzun QT sendromu (UQTS), bazal EKG'de QT aralığının uzaması ve yaşamı tehdit eden aritmiler için riski yüksek olan kalıtsal bir kalp hastalığıdır.
ORPHA: 768
Eş Anlamlısı(ları):Konjenital uzun QT sendromu
Prevalans: Bilinmiyor
Kalıtım: Otozomal dominant ya da otozomal resesif
Başlangıç yaşı: Çocukluk
ICD-10: I45.8
OMIM: 192500 220400 600919 601005 603830 611818 611819 611820 612347 612955 613485 613688 613693 613695 616247 616249
UMLS: C1141890
MeSH: -
GARD: -
MedDRA: 10057926
Özet
Epidemiyoloji
Prevalansı 2.500 canlı doğumda bire yakın olarak tahmin edilmektedir.
Klinik Tanım
UQTS’nin önemli iki belirtisi kardiyak arest ve ani kardiyak ölüme yol açabilen senkop atakları ile QT aralığının uzaması ve T dalgası anormallikleri de dahil olmak üzere elektrokardiyografik anormalliklerdir.
Etiyoloji
Hastalığın genetik temeli doksanlı yılların ortalarında tespit edildi ve şimdiye kadar tanımlanan tüm UQTS genleri, iyonik akımları düzenleyen kardiyak iyon kanalı alt birimlerini veya proteinleri kodlar. Bu genlerdeki mutasyonlar (KCNQ1, KCNH2, KCNE1, KCNE2, CACNA1c, CAV3, SCN5A, SCN4B) aksiyon potansiyelinin süresini uzatarak hastalığa neden olur. En yaygın UQTS varyantı (LQT1), KCNQ1 genindeki mutasyonlardan kaynaklanır ve genotiplendirilmiş hastaların yaklaşık yarısı KCNQ1 mutasyonları taşır.
Tanısal Yöntemler
UQTS’nin karakteristik özellikleri göz önüne alındığında, tipik vakalar hastalığın farkında hekimler için tanıda hiçbir zorluğa yol açmaz. Bununla birlikte, sınırdaki vakalar daha karmaşıktır ve spesifik tanı kriterlerinde önerildiği gibi çeşitli elektrokardiyografik, klinik ve ailesel bulguların değerlendirilmesini gerektirir. Ek olarak, moleküler tarama artık tanı sürecinin bir parçasıdır.
Yönetim ve Tedavi
Geçerli kontrendikasyonlar olmadıkça tedavi her zaman beta blokörlerle başlamalıdır. Eğer hastanın tam doz beta-blokaj olmasına rağmen ileride senkop atağı olursa, sol kardiyak sempatik denervasyon (LCSD/SKSD) olmalıdır. Tereddüt etmeksizin gerçekleştirilen ve implante edilebilen kardiyovertör-defibrilatör (KAD)ile tedavi olması, hastanın bireysel özelliklerine (yaş, cinsiyet, klinik öykü, bazı durumlarda genetik alt grubu da dahil olmak üzere mutasyona özgü özellikleri ve EKG işaretleri varlığı - 24 saatlik Holter kayıtları dahil olmak üzere) dayalı olacak şekilde nihai kararın verilmesi gerektiği düşünülmelidir.
Prognoz
Hastalığın prognozu genellikle doğru teşhis ve tedavi edilen hastalarda iyidir. Ancak hastalar için UQTS varyantı birkaç ciddi istisna vardır: (belirgin QT uzaması, 2:1 fonksiyonel atriyoventriküler blok ve sindaktili ile karakterize) Timothy sendromu, KCNQ1 mutasyonlarının taşındığı Jervell ve Lange-Nielsen sendromu (konjenital sağırlık ve kardiyak aritmilerin çok erken oluşumu ile ilişkili ciddi bir UQTS formu) ve 2:1 atriyoventriküler blok ve kardiyak aritmilerin çok erken ortaya çıktığıLQT3 hastalığı (bu terimlere bakınız)
Uzman(lar): Dr Giuseppe CELANO - Dr Lia CROTTI - Pr Peter SCHWARTZ–Son Güncelleme: Temmuz 2008
Çeviren: Ezgi Gizem BERKAY, MD
Hastalık Tanımı
Aura esnasında motor zayıflık varlığı ile karakterize nadir bir auralı migren çeşididir. Aile öyküsüne bağlı olarak iki ana formu vardır: en az bir birinci veya ikinci derece akrabasında motor zayıflığın da olduğu auralı ailesel hemiplejik migren (AHM); aile öyküsü olmayan sporadik hemiplejik migren (SHM).
ORPHA: 569
Eş Anlamlısı(ları):-
Prevalans: 1-5 / 10.000
Kalıtım: Otozomal dominant
Başlangıç yaşı: Çocukluk
ICD-10: G43.1
OMIM: 141500 602481 607516 609634
UMLS: -
MeSH: -
GARD: 1076-
MedDRA: -
Özet
Epidemiyoloji
Hemiplejik migren prevalansı 10.000'de birdir ve AHM ile SHM eşit sıklıkta görülür.
Klinik Tanım
Tipik HM atakları, en sık görülen diğer aura semptomları (her zaman duyusal, görsel ve konuşma bozuklukları) ile ilişkili olan motor zayıflık ile karakterizedir. Ek olarak, baziler tip semptomlar hastaların%70'inde görülür. Uzun süreli hemipleji, konfüzyon, koma, ateş ve nöbetlerle birlikte AHM ve SHM'de ciddi ataklar ortaya çıkabilir. Klinik spektrum ayrıca kalıcı serebellar belirtileri (nistagmus, ataksi, dizartri) ve daha az sıklıkla çeşitli nöbet tipleri ve bilişsel yetersizliği içerir.
Etiyoloji
Şimdiye kadar tanımlanan üç gen, iyon taşıyıcıları (CACNA1A, ATP1A2 ve SCNA1) kodlar. Bu üç genin taranmasıyla moleküler tanısı mümkündür.
Genetik Danışmanlık
AHM otozomal dominant kalıtımlıdır.
Yönetim ve Tedavi
Tedavi, triptanların AHM/SHM'de kontrendike olması haricinde, auralı diğer migren çeşitleri ile aynı yaklaşımları içerir. Yeni patofizyolojik bilgilere dayanarak, çeşitli antiepileptik ajanları kullanan önleyici tedaviler umut verici görünmektedir.
Prognoz
Prognoz genellikle iyidir.
Uzman(lar): Dr Anne DUCROS–Son Güncelleme: Nisan 2008
Çeviren: Ezgi Gizem BERKAY, MD
ORPHA: 444490
Eş anlamlısı(ları) : -
Prevalans:1-9/1.000.000
Kalıtım: Otozomal resesif
Başlangıç Yaşı: Bebeklik, Çocukluk, Ergenlik, Yetişkinlik
ICD-10: E78.3
OMIM: 118830 207750 238600 615947
UMLS: -
MeSH: -
GARD: -
MedDRA: -
Özet
Bu hastalık için bir Orphanet özeti halen geliştirilme aşamasındadır. Bununla birlikte, hastalıkla ilgili diğer verilere bu sayfanın altındaki Ek Bilgi menüsünden erişilebilir.
Çeviren: Ezgi Gizem BERKAY, MD
Hastalık Tanımı
Genellikle kontakt lens kullananlarda meydana gelen protozoan Akantomoeba'ya bağlı nadir görülen bir kornea enfeksiyonu, şiddetli oküler ağrı, Blefarospazm, fotofobi, göz yırtılması, bulanık görme ve yabancı cisim hissi ile karakterizedir. Derhal tedavi edilmezse, görme sorunlarına neden olabilir.
ORPHA: 67043
Eş anlamlısı(ları) : -
Prevalans: 1-9 / 100 000
Kalıtım: Uygulanabilir değil
Başlangıç Yaşı: Tüm yaşlar
ICD-10: B60.1+ H19.2*
OMIM: -
UMLS: C0000880
MeSH: D015823
GARD: 9285
MedDRA: 10069408
Çeviren: Seda KARAKAŞ, MSc - OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
Hidrojen peroksitin parçalanmasından sorumlu bir enzim olan eritrosit katalazındaki bir eksiklikten kaynaklanan nadir bir doğumsal hastalık.
ORPHA: 926
Eş anlamlısı(ları) : Katalaz Eksikliği
Prevalans: 1-9 / 100 000
Kalıtım: Otozomal resesif
Başlangıç Yaşı: Tüm yaşlar
ICD-10: E80.3
OMIM: 614097
UMLS: C0268419 C2931868
MeSH: -
GARD: 363
MedDRA: -
Özet
Epidemiyoloji
Hastalık genel popülasyonda çok nadir görülür ve 31250'de 1 olduğu tahmin edilmektedir.
Klinik Tanım
Hastalık genellikle asemptomatiktir, ancak bazı popülasyonlarda oral ülserasyonlar ve kangren veya diabetes mellitus ve ateroskleroz ile ilişkili olabilir.
Genetik Danışmanlık
Yayılımı otozomal resesiftir.
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
Klasik olarak kötü beslenme, uyuşukluk, kusma ve doğumdan kısa bir süre sonra notumda (ve daha sonra idrarda) bir akçaağaç şurubu kokusu ile karakterize nadir görülen, tedavi edilmezse ilerleyici ensefalopati ve merkezi solunum yetmezliği görülen kalıtsal bir dallı zincirli amino asit metabolizması hastalığıdır. Örtüşen dört fenotipik alt tip şunlardır: klasik, orta, aralıklı ve tiamine duyarlı AAŞH.
ORPHA: 511
Eş Anlamlısı(ları):
DZAA Eksikliği
DZAAH Eksikliği
Dallı zincirli 2-ketoasit dehidrojenaz eksikliği
Dallı zincirli ketoasidüri
AAŞH
Prevalans: 1-9 / 1 000 000
Kalıtım: Otozomal resesif
Başlangıç yaşı: Yenidoğan, erken bebeklik, çocukluk
ICD-10: E71.0
OMIM: 248600 615135
UMLS: C0024776 C0268576
MeSH: D008375
GARD: 3228
MedDRA: 10026817
Özet
Epidemiyoloji
Yayınlanmış ve yayınlanmamış yenidoğan tarama verilerine göre tahmini görülme sıklığı yaklaşık 1/150.000 canlı doğumdur.
Klinik Tanım
Klasik AAŞH, yaşamın ilk günlerinde zayıf beslenme ve uyuşukluk, ardından aralıklı apne, stereotipik hareketler ("eskrim" ve "bisiklet") ve opistotonus ile kötüleşen bir ensefalopati, 7 ila 10 gün içinde koma ve merkezi solunum yetmezliği ile kendini gösterir. Biyokimyasal testlerdeki tek anormallik ketozdur. Ara form AAŞH klinik olarak klasik AAŞH'ye benzer, ancak daha sonra başlayan ve daha az şiddetli semptomlara sahip olabilir. Araform AAŞH olguları doğumda asemptomatiktir, ancak akut dekompansasyon atakları yaşayabilir veya nörolojik semptomlar ile gelişimsel gecikme görülebilir. Tiamine duyarlı AAŞH, diyetteki lösin toleransını artıran tiamin tedavisi ile klinik olarak ara AAŞH'ye benzer.
Etiyoloji
AAŞH, dallı zincirli amino asitlerin (DZAA), lösin, izolösin ve valinin, metabolizmasında ikinci enzimatik aşamada yer alan dallı zincir 2-ketoasit dehidrojenaz (DZKAD) kompleksinin alt birimleri E1a, E1b ve E2'yi kodlayan genlerdeki mutasyonlardan kaynaklanmaktadır. DZKAD'ın dört alt birimi vardır: sırasıyla BCKDHA (19q13.1-q13.2), BCKDHB (6q14.1), DBT (1p31) ve DLD (7q31-q32) genleri tarafından kodlanan E1a, E1b, E2 ve E3. Bu genlerdeki mutasyonlar, DZAA'ların (özellikle lösin) ve bunların dallı zincirli alfa-ketoasitlerinin birikmesine yol açar. E3 alt birim genindeki (DLD) mutasyonlar AAŞH ile ilişkili değildir, ancak piruvat dehidrojenaz E3 eksikliğine yol açar (bu terime bakın). PPM1K geninde (4q22.1) bir mutasyon, tek bir hafif ara form AAŞH vakasında bildirilmiştir.
Genetik Danışmanlık
AAŞH, otozomal resesif olarak kalıtılır ve genetik danışmanlık verilebilir.
Uzman(lar): Dr Bridget WILCKEN – Son Güncelleme: Nisan 2014
Çeviren: Ezgi Gizem BERKAY, MD
Hastalık Tanımı
Rizomelia, abartılı lomber lordoz, brakidaktil ve frontal bossing ve orta yüz hipoplazisi ile makrosefali ile karakterize kondrodisplazinin bir formudur.
ORPHA:15
Eş anlamlısı(ları) : -
Prevalans: 1-9 / 100 000
Kalıtım: Otozomal dominant
Başlangıç Yaşı: yenidoğan
ICD-10: Q77.4
OMIM: 100800
UMLS: C0001080
MeSH: D000130
GARD: 8173
MedDRA: -
Özet
Epidemiyoloji
Tahmini insidans, canlı doğumda dünya çapında yaklaşık 1/25.000.
Klinik Tanım
Karakteristik klinik özellikler (rizomelia ile kısa uzuvlar, uzun ve dar gövde ve frontal boss ile makrosefali ve baskılanmış nazal köprülü orta yüz hipoplazisi) görülür. Hipotoninin yanı sıra, kısa ekstremiteler, kısa boyun ve büyük baş nedeniyle kaba motor becerilerinde başarı tipik olarak daha yavaştır. Adenoid ve tonsil hipertrofisi ile birlikte ortayüz hipoplazisi obstrüktif uyku apnesine yol açabilir. Kronik otitis media işitme sorunlarına yol açabilir. Diş çarpışıklığı yaygındır. Torakolomber kifoz bebeklik döneminde çok yaygındır. Eklemlerin çoğu hiper-uzatılabilir olabilir ve eller geniş, kısa ve çatal şeklindedir. Foramen magnum seviyesindeki kord basısı, bebeklik döneminde ve erken çocukluk döneminde santral apneye, gelişimsel gecikmeye ve uzun izlere neden olarak görülebilir. Yay bacak( Parantez bacak) sıklıkla çocuklukta görülür. Ayrıca kafa içi venöz basınç artışıyla birlikte küçük bir hidrosefali riski de vardır. Eşlik eden nörolojik eksikliklere sahip olan alt lomber spinal stenoz, erişkinlikte, kardiyovasküler hastalık gibi artmış bir sıklığa sahiptir. Obezite ortak bir konudur. Yetişkinler 131±5,6 cm (erkek) ve 124±5,9 cm (kadın) yüksekliğe ulaşır. Etkilenen kadınlar, küçük pelvis boyutundan dolayı sezaryen ile doğum yapmalıdır.
Etiyoloji
Akondroplazi, diğer fonksiyonların yanı sıra lineer kemik büyümesini düzenlemede önemli olan bir transmembran reseptörünü kodlayan fibroblast büyüme faktörü reseptörü 3 (FGFR3) genindeki mutasyonlardan kaynaklanır.
Tanısal Yöntemler
Tanı, karakteristik klinik ve radyolojik bulguların varlığına dayanır. İskelet grafileri, rizomelia, genelleştirilmiş metafizeal düzensizlikler, alt lomber vertebraların interpedik mesafesinin daralması ve küçük kare iliak kanatları ve dar sakrosiyanik çentik ile anormal bir pelvis olduğunu göstermektedir. Moleküler genetik testler, bir FGFR3 mutasyonu varlığında teşhisi doğrulayabilir.
Ayırıcı Tanı
Ayırıcı tanılar arasında hipokondroplazi, thanatoforik cücelik (tip I ve II) ve SADDAN (bu terimlere bakınız) bulunur.
Antenatal Tanı
Prenatal tanı, 3. Üç aylık dönemde rutin prenatal ultrason muayenesinde rastlantısal olarak ortaya çıkabilir. Yüksek riskli gebeliklerde veya bir ultrasondan sonra akondroplazinin şüpheli olduğu durumlarda, FGFR3 mutasyonu için tanıyı doğrulamak için fetal DNA test edilebilir. Özel laboratuarlarda implantasyon öncesi genetik tanı mümkündür.
Genetik Danışmanlık
Kalıtım otozomal dominanttır, bu nedenle genetik danışma garanti edilir. Bir ebeveynin akondroplazisi varsa, çocuğa geçirme şansı % 50'dir. Vakaların % 80'inde, ortalama boy ebeveynleri olan çocuklarda de novo mutasyon kaynaklanmaktadır. Homozigoz akondroplazi ölümcül bir durumdur.
Yönetim ve Tedavi
Yönetim çok disiplinlidir ve öngörülü bakım esastır. Bebeklerde foramen magnumun cerrahi dekompresyonu ve/veya hidrosefali için manevra gerektirebilir. Bazıları tartışmalı uzuv uzatma prosedürlerini seçebilir. Kulak enfeksiyonları ve seröz otitis medya tedavisi, işitme problemlerinin değerlendirilmesi ile birlikte gereklidir. Kaygılar ortaya çıkarsa konuşma terapisi önerilebilir. Tıkalı uyku apnesinin tedavisi adenotonsillektomi, kilo kaybı ve / veya sürekli pozitif hava yolu basıncını içerebilir. Cerrahi düzeltme bacakların bükülmesini yeniden ayarlayabilir. Yetişkin hastalarda spinal stenozu tedavi etmek için lomber laminektomi gerekebilir. Kilo alımı çocuklukta daha sonra komplikasyonları önlemek için izlenmelidir. Kraniyoservikal kavşakta yaralanma riskine neden olan faaliyetlerden kaçınılmalıdır. Sosyal ve psikolojik destek verilmelidir
Prognoz
Potansiyel olarak kardiyovasküler hastalık nedeniyle genel popülasyona kıyasla yaşam beklentisinde sadece hafif bir azalma vardır.
Uzman Hakem(ler): Dr Michael BOBER - Angela DUKER– Son Güncelleme:Nisan 2013
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
*Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
Renk körlüğü, nistagmus, fotofobi ve koni fonksiyonunun yokluğu veya bozulması nedeniyle ciddi derecede azalmış görme duyarlılığı ile karakterize nadir görülen bir otozomal resesif retinal bozukluktur.
ORPHA:49382
Eş anlamlısı(ları) : ACHM, Kısmi ya da tamamen renk körlüğü, Pingelapese körlük, Çubuk Monokromasi, Çubuk monokromatizması, Toplam renk körlüğü
Prevalans: 1-9 / 100 000
Kalıtım: Otozomal resesif
Başlangıç Yaşı: Yenidoğan, bebeklik
ICD-10: H53.5
OMIM: 216900 262300 610024 613093 613856 616517
UMLS: C0152200
MeSH: -
GARD: -
MedDRA: 10000454
Özet
Epidemiyoloji
Prevalansın dünya çapında 1/30.000-1/50.000 olduğu tahmin edilmektedir.
Klinik Tanım
ACHM, azalmış görme duyarlılığı, sarkaç nistagmus, ışığa duyarlılık (fotofobi), küçük bir merkezi skotoma ve azalmış veya tamamen renk ayırtetme kaybı ile karakterize edilir. Çoğu birey, üç koni türünün tamamında işlev eksikliği olan tam ACHM'ye sahiptir. Nadiren, bireyler benzer, ancak genellikle daha az şiddetli semptomları olan eksik ACHM'ye sahiptir.
Etiyoloji
Beş gen (GNAT2 (1p13), PDE6C (10q24), PDE6H (12p13), CNGA3 (2q11.2) ve CNGB3 (8q21.3)), koni fototransdüksiyon kademeleri (G kondansatörünün tüm ana bileşenlerini kodlayan) -protein GNAT2> fosfodiesteraz PDE6C/PDE6H> siklik nükleotit kapılı kanal CNGA3/CNGB3) ACHM ile ilişkilendirilmiştir. CNGB3'teki mutasyonlar en yaygın olanıdır, bunu CNGA3 izler, diğerleri nadir görülen ACHM nedenleridir.
Tanısal Yöntemler
ACHM tanısı, fotopik fakat normal skotopik cevapların kaybının gözlendiği klinik oftalmolojik muayene, psikofiziksel test (yani renk görme) ve elektrofizyolojik teste (elektroretinografi - ERG) dayanmaktadır. Optik koherens tomografi, fotoreseptörlerin iç/dış segment kavşağında progresif bozulma ve / veya kayıp ve maküler bölgede retinal pigment epitelinin (RPE) zayıfladığını gösterir. Teşhis, nedensel genlerin moleküler genetik analizi ile doğrulanır.
Ayırıcı Tanı
Ayırıcı tanı, mavi koni monokromatizmi (BCM), Leber konjenital amaurozu, diğer koni distrofileri (bu terimlere bakın) ve serebral akromatopyayı içerir.
Antenatal Tanı
Riskli çiftlere uzman laboratuvarlar tarafından doğum öncesi tanı önerilebilir.
Genetik Danışmanlık
ACHM, otozomal resesif bir şekilde iletilir. Ailede mutasyon riski taşıyan taşıyıcı testleri ailede hastalığa neden olan mutasyonlar tanımlandıktan sonra mümkündür. Ayrıca, risk altındaki çiftlere (her iki birey de hastalığa neden olan bir mutasyonun taşıyıcısıdır) genetik danışmanlık verilmelidir, bu da etkilenen bir çocuğa sahip olma şansı% 25'tir.
Yönetim ve Tedavi
Spesifik bir terapi yok. Yönetim semptomatiktir ve düzenli oftalmolojik takip muayenesini içerir. Hastalara fotofobiyi azaltmak ve kontrast duyarlılığını arttırmak için filtreleme gözlükleri veya kontakt lensler (kırmızı renkli veya kahverengi) kullanma olasılığı hakkında bilgi verilmelidir. Az-gören yardımlar, okuma için yüksek güçlü büyüteçleri içerir.
Prognoz
ACHM genellikle durağan bir hastalıktır, ancak maküler dejenerasyon oluşabilir.
Uzman Hakem(ler): Dr Susanne KOHL– Son Güncelleme:Ağustos 2013
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalığın Tanımı
Akromegali, aşırı büyüme hormonu (GH-growth hormone) üretiminin sebep olduğu kazanılmış bir bozukluktur. Özellikle yüz ve ekstremitelerde belirgin olmak üzere progresif şekil bozukluğu ve sistematik belirtilerle karakterizedir.
ORPHA:963
Hastalığın diğer adı/adları: -
Görülme Sıklığı: 1-9 / 100 000
Kalıtımı: Yoktur
Görülmeye başladığı yaşlar: Yetişkinlik
ICD-10: E22.0
OMIM: 102200 300943
UMLS: C0001206
MeSH: D000172
GARD: 5725
MedDRA: 10000599
Özet
Epidemiyoloji
Avrupa’daki görülme sıklığının 1/250,000-1/100,000 olduğu tahmin edilmektedir. Çoğu zaman orta yaştaki erişkinlerde tanı koyulur (ortalama 40’lı yaşlarda, kadınlar ve erkekler eşit ölçüde etkilenir).
Klinik Açıklaması
Sinsi bir şekilde başlaması ve yavaş ilerlemesi nedeniyle tanının konması hastalığın başlangıcından itibaren 4-10 yıl veya daha fazla sürebilir. Başlıca klinik belirtiler büyümüş ekstremiteler (eller ve ayaklar), genişlemiş, kalınlaşmış ve güdükleşmiş parmaklar ve yumuşak dokuda kalınlaşmadır. Yüz görünümü karakteristiktir; genişlemiş ve kalınlaşmış bir burun, belirginleşmiş elmacık kemiği, çıkıntılı alın, kalın dudaklar ve belirgin yüz çizgileri görülür. Alın ve üzerindeki cilt kalınlaşmıştır, bazen frontal çıkıntı oluşumuna yol açar. Prognatizm, maksiller genişleme, diş ayrılması ve çene malokluzyonu ile birlikte mandibulanın aşırı büyümesine doğru bir eğilim vardır. Hastalığın ayrıca romatolojik, kardiyovasküler, respiratuar ve metabolik sonuçları vardır ve prognoz açısından belirleyidir.
Etiyoloji
Olguların büyük kısmında, akromegali hipofiz bezi tümörüne bağlıdır; bu saf büyüme hormonu sentezleyen (60%) veya mikst tip olabilir. Çok nadir vakalarda akromegali, büyüme hormonu salgılayıcı hormonun (GHRH) ektopik olarak sentezlenmesinin sorumlu olduğu hipofiz bezi hiperplazisine bağlıdır. Aril hidrokarbon reseptörü etkileşimli protein geni, AIP (11q13.3), özellikle ailevi olgularda ya da akromegalinin çocukluk veya adolesan çağda başladığı durumlarda ana yatkınlık faktörü olarak tanımlanmıştır. Akromegali ayrıca MEN1 (MEN1 geni, 11q13) veya Carney kompleksi (PRKAR1A geni , 17q24.2) (bu terimlere bakınız) gibi çoklu endokrin neoplazi (MEN) sendromlarının parçası olabilir.
Tanı yöntemleri
Klinik tanı, oral glikoz tolerans testi (OGTT) sonrasında artmış serum GH konsantrasyonuyla ve artan insülin benzeri büyüme faktörü-I (IGF-I) seviyelerinin tespiti ile biyokimyasal olarak doğrulanır. Tümörün boyutu ve yaygınlığı görüntüleme yöntemleriyle tespit edilir. Ekokardiyografi ve uyku apnesi testi, akromegalinin klinik etkilerini belirlemede kullanılır.
Yönetim ve tedavi
Tedavide amaç tümörün bası yapıcı etkisini hastalık yapıcı lezyonu ortadan kaldırarak düzeltmek (veya önlemek) ve GH ile IGF-I seviyelerini normale düşürmektir. Transsfenoidal cerrahi genellikle ilk seçenektir. Ameliyat sonrası GH / IGF-I hipersekresyonu normalleşmezse, dopamin agonistleri ve / veya somatostatin analogları ile tıbbi tedavi önerilmektedir. Somatostatin analoglarına dirençli hastalarda GH antagonisti (pegvisomant) kullanılır. Medikal tedavi başarısız olduğunda radyoterapi seçenek olarak sunulabilir.
Prognoz
Hormonal bozukluğun etkin kontrolü birçok vakada sağlanmakta, yaşam beklentisi genel populasyon düzeyine çekilebilmekle birlikte kür veya etkin kontrol sağlansa bile, çoğunlukla sekel (eklem ağrısı, deformiteler ve hayat kalitesinde bozulma) kalmaktadır.
Uzman hakem(ler): Pr Philippe CHANSON - Dr Sylvie SALENAVE – Son güncelleme: Ocak 2014
Çeviren: Ayşe Nur Kurtcuoğlu – OCAK 2019
Kaynak sayfa için tıklayınız
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
ORPHA:178320
Eş anlamlısı(ları) : -
Prevalans: 1-5 / 10 000
Kalıtım: -
Başlangıç Yaşı: -
ICD-10: -
OMIM: -
UMLS: C0242488
MeSH: D055371
GARD: -
MedDRA: 10069351
Özet
Bu durum, Avrupa ve/veya Amerika’da yetim olarak tanımlanan nadir bir durumdur. Bu terim farmakogenetik test için endikasyon olarak belirtilmiştir.
Çeviren: Ezgi Gizem Berkay, MD – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
Santral sinir sisteminin demiyelinizan bir bozukluğudur.
ORPHA:83597
Eş anlamlısı(ları) : ADEM, Akut dissemine ensefalit
Prevalans: Bilinmiyor
Kalıtım: Uygulanabilir değil
Başlangıç Yaşı: Tüm yaşlar
ICD-10: G04.0
OMIM: -
UMLS: C0014059
MeSH: -
GARD: 8639
MedDRA: -
Özet
Epidemiyoloji
İnsidans oranı yılda 125.000-250.000 arasında 1 olarak tahmin edilmiştir.
Klinik Tanım
10 yaşından küçük çocuklar ağırlıklı olarak etkilenir. Mevsimsel kış ve ilkbahar zirveleri kaydedilmiştir. Hastalık genellikle akut viral veya bakteriyel enfeksiyon veya aşılamadan sonra, ani bir irritabilite başlangıcı ve 1-4 haftalık bir prodromal sürenin ardından uyuşukluktan sonra gelişir. Başlıca belirtiler ateş, baş ağrısı, uyuşukluk, zihinsel durumdaki değişiklikler, nöbetler ve komadır. Zayıflık, kusma, kilo kaybı, boyun boynu, ataksi, bilateral optik nörit ve deliryum yaygındır. Literatür verilerine göre periferik sinir sistemi tutulumu (tek bacak veya hemiplejik felç) olguların% 5-45'inde görülür. Başlangıçta ADEM semptomları hafif olsa da, birkaç saat ila dört gün içinde hızla kötüleşir. Patolojik olarak ADEM, serebral ve serebellar beyaz maddede iki taraflı büyük ve birleşik lezyonlarla karakterize edilir; Bazal ganglionlar ve gri madde de dahil olabilir. Omurilikteki ADEM lezyonları süreklidir ve çok sayıda seviyeye uzanır. Bazı yazarlar hastalığı, multipl sklerozun (MS) bir varyant veya sınır çizgisi formu olarak görür (bu terime bakınız).
Etiyoloji
ADEM, merkezi sinir sisteminin immün aracılı bir bozukluğu olarak kabul edilir. Kendiliğinden ortaya çıkabilir, ancak çoğu durumda hastalık bulaşıcı hastalık veya aşılarla tetiklenir. Pasteur kuduz aşısı ile ADEM arasındaki bağlantılar belgelenmiştir. ADEM ile daha az ilişkilendirilen aşılar arasında boğmaca, kızamık, Japon B virüsü, tetanoz, grip vardır.
Tanısal Yöntemler
Teşhis klinik öykü ve tercih edilen tanı yöntemi olan manyetik rezonans görüntülemeye dayanmaktadır.
Ayırıcı Tanı
MS ana ayırıcı tanıdır. ADEM ile MS'in ilk bölümü arasındaki ayrım çok zor olabilir ancak önemli prognostik ve tedavi sonuçları vardır. Ayırıcı tanı ayrıca enfeksiyöz ensefalit, Guillain-Barré sendromu, glioblastoma multiforme, Schilder hastalığı (bu terimlere bakınız), akut başlangıçlı psikotik bozukluklar, toksik / metabolik ensefalopati, vaskülit, vaskülitik olmayan otoimmün ensefalopati, menenjit, metastatik hastalıkları içerir.
Yönetim ve Tedavi
Günümüzde akut olaylar için kortikosteroidlerle immünsüpresyon tedavinin temelidir. Steroid tedavisine dirençli hastalarda, plazmaferez veya intravenöz immünoglobulin tedavisi kullanılabilir. Semptomatik ve destekleyici tedaviler önerilir.
Prognoz
ADEM tipik olarak, genellikle olumlu bir sonuç veren monofazik (tek bölüm) bir hastalıktır. Ortalama iyileşme süresi bir ila altı ay arasındadır. Tekrarlayan vakaların% 5-25'inde, genellikle başlamasından 6-18 ay sonra ortaya çıkabilir. Akut beyin ödemi görülen olguların çok azında yoğun bakım gereklidir.
Son Güncelleme:Temmuz 2009
Çeviren: Seda KARAKAŞ, MSc – OCAK 2019
Kaynak sayfa için tıklayınız.
Not: Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalık Tanımı
Guillain-Barré sendromunun (GBS) klinik spektrumuna ait nadir bir enflamatuvar nöropatidir.
ORPHA:98916
Eş Anlamlısı(ları):
AIDP
Akut İdiyopatik Demiyelinizan Polinöropati
Akut İnflamatuvar Polinöropati
GBS, Akut İnlamatuvar Demiyelinizan Poliradikülonöropatik Form
Guillain-Barré Sendromu, Akut İnfalamatuvar Demiyelinizan Poliradikülonöropatik Form
Prevalans: 1-9 / 100 000
Kalıtım: Multigenik/Multifaktöriyel ya da Uygulanamaz
Başlangıç yaşı: Tüm yaşlar
ICD-10: G61.0
OMIM: 139393
UMLS: C1963929
MeSH: -
GARD:-
mMedDRA: -
Özet
Epidemiyoloji
Genel GBS insidansının 1/91.000 ila 1/55.000 arasında olduğu tahmin edilmektedir. AIDP, Avrupa ve Kuzey Amerika'daki GBS vakalarının yaklaşık %90'ını oluşturmaktadır ve bu nedenle GBS terimi Batı ülkelerinde genellikle AIDP ile eşanlamlıdır. Hastalık her yaştaki hastalarda görülür ve erkekler kadınlardan yaklaşık 1,5 kat daha fazla etkilenir.
Klinik Tanım
AIDP'nin klinik seyri üç aşamaya ayrılmıştır. İlk aşama (birkaç hafta süren), hızlı ilerleyen kas güçsüzlüğü ile karakterizedir (genellikle önce ayaklarda ortaya çıkar ve yukarı doğru ilerler). Simetriktir ve akut nöromüsküler paraliziye neden olabilir. Duyusal bozukluklar (karıncalanma ve uyuşukluk), yoğun ağrılar ve kramplar da ortaya çıkabilir. Tutulan diğer alanlar, solunum kaslarını (akut solunum yetmezliğine yol açan,% 20-30'unun mekanik ventilasyona ihtiyacı olan), yutma kaslarını (hayatı tehdit edici aspirasyona neden olan) ve göz kaslarını (oftalmoplejiye neden olan) içerebilir. Derin tendon refleksleri azalmış veya yok olabilir. İkinci aşamada (değişken süre), semptomlar stabil hale gelir, ancak diğer belirtiler (kardiyak aritmi, hiper / hipotansiyon ve gastrik dismotilite) oluşabilir. Üçüncü (iyileşme) aşamasında, birkaç ay veya daha uzun süren semptomlar yavaşça geriler. Birçok hasta aylarca hatta yıllarca rezidüel bulgulara sahiptir (zayıflık, duyusal bozukluklar, yorgunluk veya ağrı).
Etiyoloji
Vakaların çoğunda, enfeksiyöz bir hastalık, ekstremite güçsüzlüğünün başlamasından önce gelir ve Campylobacter jejuni enfeksiyonu en sık görülenidir. Sitomegalovirüs, Epstein-Barr virüsü, Mycoplasma pneumoniae ve Haemophilus influenza da bu duruma yol açmaktadır. AIDP ayrıca aşılama ve cerrahi müdahale sonrası da rapor edilmiştir. Patolojik mekanizmalar araştırılmaya devam etmesine rağmen, AIDP, miyelin kılıflarının aktive edilmiş makrofaj infiltrasyonu ile ilişkili olup, miyelin hasarına ve demiyelinasyonuna neden olmaktadır. Diğer immünolojik mekanizmaların da rol oynaması muhtemeldir.
Tanısal Yöntemler
Teşhis klinik tabloya dayanır ve tanı koymak zor olabilir. Lomber ponksiyon yapılarak beyin omurilik sıvısı (BOS) incelenmelidir ve elektromiyografi, GBS tanısında ve alt tipinin tanımlanmasında yardımcı olabilir: AIDP veya aksonal (AMAN, AMSAN) formları (bu terimlere bakın).
Ayırıcı Tanı
Ayırıcı tanı ilaca bağlı nöropati, kritik hastalık polinöropatisi, karsinomatozis, metabolik bozukluklar, akut rabdomiyoliz ve omurilik ve spinal sinir kökü sıkışması veya enflamasyonunu içerir. Ayırıcı tanıda porfiri, vaskülit, difteri, myastenia gravis, botulizm, polimiyozit, dermatomiyozit, beyin sapı ensefaliti, menenjit, transvers miyelit ve poliomyelitin (bu terimlere bakınız) yanı sıra B1 vitamini eksikliği de düşünülebilir.
Yönetim ve Tedavi
Yoğun bakım olanakları sağlayan multidisipliner bir ekibin optimal bakımı, hastalığın yönetimi için şarttır. Tedavi, intravenöz immünoglobülin (IVIg) veya plazma değişiminin hızlı uygulanmasından oluşur. Fizyoterapi ve rehabilitasyon da önemlidir.
Prognoz
GBS hastalarında değişken prognoz vardır: hastaların yaklaşık %50'sinin tamamen düzeldiği ya da sadece küçük sekellerin kaldığı, %20'sinin 6 ay sonrasında yürüyemediği ve %3'ünün öldüğü tahmin edilmektedir. Çok sayıda klinik, elektrofizyolojik, serolojik ve laboratuvar faktörün olması, olumsuz sonucun göstergesi olarak tanımlanmıştır. Yorgunluğa ve dayanıklılığa tahammülsüzlük yıllarca sürebilir.
Uzman(lar): Pr P.A. [Pieter] VAN DOORN – Son Güncelleme: Aralık 2009
Çeviren: Ezgi Gizem BERKAY, MD – OCAK 2019
Kaynak sayfa için tıklayınız.
Not :Bu sayfada yer alan kayıtlar, uluslararası OrphaNet web sitesinden tercüme edilmiş olup, okuyucuyu çeşitli nadir hastalıklar hakkında tamamen genel bir fikir sahibi yapmaya yönelik olarak hazırlanmıştır. Veriler güncel olmayabilir. Tıbbi bilgi niteliği taşımamakta olup, tanı, tedavi veya herhangi bir klinik karar verme sürecinde kullanılmamalıdır.
Hastalarımızdan gelen sorular ve cevapları
Sitemizin sayın ziyaretçileri,
Sitemiz aracılığıyla tarafımıza çok yoğun miktarda başvuru yapılmakta ve soru sorulmaktadır. Bu sebeple tüm sorulara yanıt veremeyeceğimiz için sık sorulan bazı soruları derleyerek bu bölümde yayınlamayı uygun gördük. Bu bölümde aradığınız yanıtı bulamadığınız takdirde bizimle temasa geçmekten çekinmeyiniz.
Saygılarımla,
Prof.Dr.Buğra HARMANDAR
Doğumsal kalp hastalığı olan çocuğunuzla ilgili olarak daha detaylı bilgi almak veya kalp hastalığı nedeniyle ameliyat planlanan çocuğunuz hakkında görüşmek üzere
Muayene ve Randevu Tel:
0.553.7665470 (GSM)
Hastane:
0.252.2141323 veya
0.252.2141324 'den
dahili 4367 veya 3696 veya 3695 no’lu telefonları tuşlayarak randevu alabilir ve"Kalp ve Damar Cerrahisi Anabilim Dalı Başkanı"Prof.Dr.Buğra HARMANDAR ile yüzyüze görüşebilirsiniz.
Sorular ve Cevaplar
Soru 1. Merhaba bugün 41 günlük olan bi erkek bebeğim var bebeğim doğduğunda DORV,VSD,Pulmoner Atrezi ,PDA Tanısı koyuldu. Kalp boşlukları normalden geniştir IVS'de VSD vardır Aorta ve pulmoner arter sağ ventrikülden çıkmaktadır.,aorta sağdadır .Pulmoner kapak atretiktir,ana PA ve dalları iyi gelişmiştir,duktal akımla dolmaktadır,Arkus sağ seyirlidir koarktasyon yoktur. Tanısı ve yorumu yukardaki gibi olan oğlumun hastalığında anjiyo olması gerektiği söylendi adanadaki dr 6aylıkken bursadaki dr 3 aylıkken olması gerektini söylediler şant ameliyatına gerek görmediler sizce anjiyo ve ameliyat için doğru tarih nedir teşekkürler şimdiden
Cevap: Böyle bir çocuğu 3 ay - 6 ay bekletmek doğru değil. PDA kapanabilir ve akciğer damarlarına kan akışı durabilir. Hastayı en kısa zamanda hastanemize veya bir pediatrik kardiyoloji ünitesine götürmenizi tavsiye ederim.
Soru 2. 45 aylık kızımda VSD teşhisi 15 günlükken konuldu ve o günden bu yana düzenli olarak kontrol altında. ilk 14 ay enapril, digi ve lasix kullandık ancak daha sonra hiç bir ilaç kullanmadık. ilaçları kullandığımız süre içerinde ciddi kilo sorunumuz vardı, (18 aylıkken 7,5 kg dı) ancak boy uzaması devam ettiği için ve doğum kilosu da 2,5 kg olduğundan herhangi bir müdehaleye gerek görülmedi ve doğal akışına bırakıldı. ama daha sonrasında boy ve kilomuz normale geldi yavaş yavaş. şimdi 14 kg ve 98cm. boyunda. kızım bu süreçte deliğini kapatmaya çalışmakla birlikte, henüz başaramadı ve aort yetmezliği gündeme geldiği için şimdi ameliyat planlanıyor. bu ameliyatlarda süre nedir ve nasıl işler. ameliyat sonrasında bizi neler bekliyor. bu ameliyatlar nerelerde yapılıyor, ne kadar sürede ameliyata hazırlanılabiliyor. ameliyata karar verildiğinde hemen ameliyat edilebiliyormu?
Cevap: VSD'ler doğumdan itibaren tespit edilebilmekle birlikte hepsine acil ameliyat gerekmemektedir. Küçük VSD'ler önemsiz olup ömür boyu ameliyat gerektirmeyebileceği gibi büyük VSD'ler hayatın ilk yılı hatta ilk ayları içerisinde ameliyat gerektirebilir. Sizin kızınızda olduğu gibi aort kapak yetersizliğine yol açabilen subaortik yani aort kapak altında yer alan VSD'ler eğer küçük ise ve aort kapak yetersizliğine yol açmıyorsa hasta büyüyünceye kadar beklenebilir. Düzenli eko takipleri ile kontrol edilen bu VSD'lerde eğer aort kapak yetersizliği oluşmaya başlarsa en kısa zamanda ameliyat edilmelidirler. Çünkü bozulan bir aort kapağın geri dönüşü yoktur ve ileride aort kapağın mekanik bir kapakla değiştirilmesini gerektirebilecek ameliyat ihtiyacı gösterebilirler. Dolayısı ile kızınızın artık cerrahi müdahaleye ihtiyacı olduğunu ve beklemenin gereksiz olduğunu söyleyebiliriz. Bu safhadan sonra delik artık kendi kendine de kapanamaz. Bu tür deliklerin cerrahi olmadan anjio ve katater ile kapatılması da mümkün değildir. Bu ameliyatlar çocuk kalp cerrahisi yapılan pekçok hastanede yapılabiliyor. Fakat yine de bu hastaneler arasında tecrübesi en fazla olan ve yıllık ameliyat sayısı en fazla olanları tercih etmekte fayda var. Bu açıdan yıllık 750 çocuk açık kalp ameliyatı ile 2012 yılında Young Investigation Award ödülünü almış bir merkez olarak size Dr.Siyami Ersek Hastanesini tavsiye edebilirim. Hastaların ameliyata hazırlığı yalnızca birkaç gün sürmekle birlikte ameliyat sonrası hastanede kalış süremiz ortalama 1 haftadır. Ameliyat için bekleyen çok sayıda hastamız olmasına rağmen elimizden geldiğince hızlı sürede bekleyen hastalarımıza yardımcı olmaya çalışıyoruz. Eğer bizimle temas kurarsanız size yardımcı olacağız.
Soru 3. Bir aylık kızımın çekilen ekosunda 4 mm ve 1 mm olmak üzere iki asd,perimembranöz outlet-inlet geniş bir adet vsd görüldüğü,iki venrtrikül arasında basınç farkının olmadığı,kapakların,arterin normal olduğu arkus aorta solda yerleşimli olduğu aort koarktasyon görülmediği belirtilmiştir. Sormak istediğim ameliyatın riskli olup olmadığı,ameliyatın açık kalp dışında başkaca yöntemle kapatılma imkanı olup olmadığı,şuan itibariyle bebeğin hayati riskinin olup olmadığıdır.
Cevap: 10 yıllık vaka taramalarımızda 1 yaş altı geniş VSD'li hastalarda ameliyat sonrası ölüm oranımız % 1,4'tür. Yaptığımız çok daha kompleks ameliyatlar yanında nispeten riski daha düşük bir ameliyattır. Ama her açık kalp ameliyatında olduğu gibi bu ameliyatta da bir açık kalp ameliyatının taşıdığı riskler mevcuttur. Ameliyat dışında anjio ile kapatma yöntemi var fakat yalnızca muskuler VSD'ler bu yöntemle kapatılabiliyor. Geniş perimembranöz bir VSD'nin anjio katater ile kapatılma imkanı yok. Ancak ameliyatla kapatılabilir. VSD dışında ASD'lerinin hiçbir önemi yok. VSD kapatılırken 4 mm olan asd'yi de basitçe kapatmak mümkün. Eğer bebeğin kilo alımı yeterli ise ve aylık eko kontrollerinde pulmoner hipertansiyonu ilerlemiyorsa (akciğer damar basıncı) 1 yaşına kadar beklenebilir. Ama biz bu tür hastalarda ilk 6 ay tercihen ilk 3 ay içerisinde vsd'yi kapatmayı tercih ediyoruz. VSD'nin erken kapatılması hastada kalıcı hipertansiyon gelişimini önlediği için ilerideki yaşamını da rahat geçirmesini sağlayacak en iyi tedbirdir. Bu bilgiler dışında çocuğun şu haliyle çok acil bir hayati tehlikesi bulunmamaktadır. Eğer bizimle irtibata geçerseniz yardımcı olacağız.
Soru 4. Öncelikle böyle bir forum kurduğunuz için çok teşekkürler .on aylık olan oğlum vsd + pa +mapca teşhisiyle yedi ay önce farklı bir hastanede şant ameliyatı oldu.Şantın yeri ameliyat raporunda belirtilmemiş.Ekolarda şant akımı görülmüyor.Şant bulunamıyor.Rpa :5 Lpa :5,4 desenden aorta 3,5 mm lik mapcası mevcut .Tekrar şant ameliyatına gerek görülebilir deniyor.Ama bu olanlardan sonra güvenimiz kırılmış durumda .Öneriniz ne ?
Cevap: Öncelikle geçmiş olsun. VSD+PA+MAPCA'lı hastalar nativ (doğal) pulmoner arterleri iyi gelişmemiş bir hasta grubu olup bunların şant ameliyatlarında sorun olabilmektedir. Hastaya ikinci bir şant ameliyatı yapılabilir. Başvurmanız halinde yardımcı olmaya çalışacağız.
Soru 5. 21 haftalık yapılan detaylı usg sinde Kalp: Sol Ventrikül içi 1,7 mm Hipereko mevcuttur. Bu sonuca binaen yapılan amino sentez sonucunda DOWN Sendromu ile alakalı genetik bir faktörün olmadığı tespit edilmiştir.Çekilen detaylı Fetal EKO raporunda ise - AV Kanal Defekti - Tek AV Kapak ve Yetersizliği - Primum Atrial Septal Defekt - İnlet Ventriküler Septal Defekt sonuçlarına varılmış olup; bu aşamadan sonra yapılabileceklerimizin neler olduğu konusunda görüş ve önerilerinizi bekliyoruz. Not: Gebelik bu tarih itibariyle 23 H 1 G lüktür.
Cevap: AV Kanal Defekti - Tek AV Kapak ve Yetersizliği - Primum Atrial Septal Defekt - İnlet Ventriküler Septal Defekt tanısına sahip hastaların büyük çoğunluğu down sendromlu olmakla beraber down sendromu olmayanlara da rastalanabilmektedir. Bu hastaları yaklaşık 3-4 aylık oluncaya kadar medikal tedavi ile takip ediyor ve ardından ameliyata alıyoruz. Bu konuyla ilgili 10 yılın üzerindeki deneyimlerimizi anlatan ve SCI-e kapsamında yayınlanan yazımızın linkini aşağıda veriyorum.
Article: Results for surgical correction of complete atrioventricular septal defect: associations with age, surgical era, and technique.
Bugra Harmandar, Numan Ali Aydemir, Ali Riza Karaci, Ahmet Sasmazel, Turkay Saritas, Mehmet Salih Bilal, Ibrahim Yekeler
Journal of Cardiac Surgery 11/2012; 27(6):745-53.
Soru 6. Oğlum vsd pa mapca tanısıyla bir yıl önce şant ameliyatı oldu.rpa 4mm lpa 4mm idi.şuan so2 78 rpa 6,5 lpa 6,5 dsaorta 10,5 .kilo 10,5 .aortta 3,5 mm lik mapcası var .kalp yetmezliği bulgusu yok .nasıl bir yol izlenmeli.damar gelişimi normal mi? Şuan 14 aylık.
Cevap: Çocuğunuzun her iki pulmoner arterinde de bir miktar büyüme olması sevindirici. Saturasyonu sınırda olduğu için bir miktar daha beklenenilir ama 70-75 değerlerinin altına düştüğünde hala pulmoner arterleri bu sınırlarda ise yeni ve daha büyük bir şanta ihtiyacı olacak gibi gözüküyor. Çünkü pulmoner arter gelişimi henüz tam düzeltme ameliyatına yetecek kadar değil. Basitçe rpa + lpa / desendan aorta = 1,7 - 1,8 değerlerinin üzerinde olmasını yeterli olarak kabul ediyoruz. Eskiden bu oran 2 civarındaydı. Ama yine de vsd pa mapca tanısı olmasına rağmen gerçek pulmoner arterlere sahip olması da hasta açısından iyi bir durum. Gerçek pulmoner arterleri olmayan hastalar da olabiliyor ve bu hastaların seyirleri daha kötü oluyor. Geçmiş olsun.
Soru 7. Damarları ameliyat yapılacak düzeye gelirse homogreftin avantajı olur mu? Temini nasıl olur ?
Cevap: Homogreft avantajlı olur. Biz de homogreftleri sıkça kullanıyoruz. Son 1 yıldır temininde güçlük çekmiyoruz. Ticari olarak bize SGK ödemesi ile birlikte teslim ediliyor. Aileler herhangi bir ücret ödemiyor.
Soru 8. 20.08.2013 te bir oğlum oldu ve AVSD teşhisi konuldu. erzurumda götürdüğümüz araştırma hastanesi ameliyat edilmesi gerektiğini söyledi. ilk 6 ay içinde olmalı dediler. araştırdım ve hastanenizin bu konuda eniyi hastane olduğunu gördüm. lütfen bu konuda bana yardımcı olabilirmisiniz. ne zaman ameliyat yapılmalıdır. ne gibi tavsiyeleriniz olur. Lasix ve başka ilaçlar verdiler bir hap ve bir damla. bunları çöcoğa verdiğimizde çocuk kusuyor. bu konuda ne yapalım. bu ilaçlar neişe yarar ve kullanmaz isek ne gibi sorunlar olur.
Ayrıca AVSD ile Down sendromu arasında bir ilişki varmıdır, çocuğun yüzü sendromlulara benziyor ve genetik test için kan verdik. bu konuda bizi aydınlatabilirmisiniz ?
Cevap: Avsd sık yaptığımız ameliyatlardan olup türkiye genelinde bu konuda en fazla ameliyatı yapan ve sonuçları en iyi olan ekibiz (%2-3 ölüm oranı ile). Avsd ameliyatlarında en iyi sonuçlar ilk 6 ay içerisinde yapılan ameliyatlarda alınmakra olup yaptığımız çalışmalarda 8 aya kadar iyi sonuçların alınabileceğini gösterdik. Kalbin içerisindeki delik çok büyük olduğu için buradan akciğere giden aşırı kan akciğer damar duvarlarını zamanla sertleştirerek pulmoner hipertansiyon denen hastalığa yol açar. Eğer tedavi edilmezse 1 yaşından sonra pulmoner hipertansiyon geri dönüşümsüz olur ve hasta ameliyat şansını kaybeder. Ameliyat ilk 6 ay içerisinde yapılmalıdır fakat yaptığımız çalışmalarda 4 ay altında ameliyat edilenlerin kalp kapaklarında kaçak miktarının daha fazla olduğunu gördük. O yüzden 4-6 ay arası ameliyat önermekteyiz. Bu çocukların akciğerleri bozuk olduğu için devamlı akciğer enfeksiyonuna adaydırlar. O yüzden hastaları enfeksiyondan ciddi şekilde korumak gerekir. Çünkü akciğer enfeksiyonu varken ameliyat yapamıyoruz. Ve tekrarlayan akciğer enfeksiyonları nedeniyle hastalar 6 ay sınırını aşabiliyor ve ameliyat şansını kaçırabiliyor. Çocuğu insanlarla temastan uzak tutun, özellikle kimse öpmesin. Verilen ilaçları kullanmanız şarttır. Kusmaması için sulandırarak yavaş yavaş ve birbirinden ayrı zamanlarda verin. Kullanmazsa kalp yetersizliğine girebilir. Bu hastaların %85'i down sendromudur. Down olmayanlar genelde parsiyel avsd'dir ve acil ameliyat gerekmez, ilk 5 yaşta ameliyat edilir.
Soru 9. Öncelikle doğacak olan 21 haftalık bebeğimize konulan "ARSA" - "Vasküler Ring" yani bize iletilen hali ile "kalpten sağ kola giden damarın soluk borusunun önü yerine arkasından gitmesi" konusunda araştırma yaparken
www.pediatrikkalpcerrahisi.com adresinden mail bilginize ulaştık. Bilgi desteği ricamızı umarım anlayışla karşılarsınız. ARSA ile ilgili kendi doktorumuzun ilettikleri dışında ekstra bir bilgi, yapabileceğimiz bir şey var mı diye araştırıyoruz. ARSA'nın ne olduğu ve çıkarabileceği sorunları öğrendik. Bize verilen bilgi dahilinde down sendromu riski için ambiyosentez de yaptırdık ve sonucunu bekliyoruz. Aklımıza takılan soruyu size yöneltmek istiyoruz. Doktorların konu hakkında ilettiği farklı bilgiler mevcut. Bazı doktorlar "ARSA bir sorun teşkil etmez ve yaşamda zorluk çıkarmaz" derken, internette ise bu konuda yapılan ameliyatlara rastlamak mümkün. Benim anladığım, ARSA'nın hayatın belli bir döneminde çok büyük oranda semptomlara yol açtığı yönünde. Bu doğru mudur acaba, bebeğin hayatı boyunca bir sorunla karşılaşmadan yaşamını geçirmesi çok düşük bir olasılık mıdır yada mümkün değil midir. Biz bebek dünyaya geldiğinde doğar doğmaz ekokardiyografi ve gerekirse MR Anjiyo yaptıracağız. Bunlarda bir sorun gözükmese dahi cerrahi müdahale seçeneğini değerlendirmeli miyiz sorusunun yanıtı maalesef bizde net değil. Bebekken olmayan semptomların çocukluk çağında çıkması halinde ameliyat sanırım daha zor olur. Bir taraftanda "ya hayatı boyunca semptom vermez ise boşuna ameliyat ettirmiş olur muyuz" sorusu aklımızda. Bizi bilgilendirme ve yönlendirme nezaketinde bulunursanız çok memnun oluruz ?
Cevap: Aberran sağ subklaviyan arter inkomplet vasküler ringler grubunda yer alır. Bu hastalarda sağ kola gitmesi gereken subklaviyan arter aortun ilk dalı olan truncus brachiocephalicustan çıkıp sağ kola gideceği yerde aortun sol tarafından son dal olan sol subklaviyan arterden çıkarak, sol taraftan sağa doğru gider. Soldan sağa doğru seyri sırasında trakea (soluk borusu) ve özefagus (yemek borusu)'un genellikle arasından bazen de her ikisinin de arkasından geçerek bu yapılara çeşitli derecelerde bası semptomları oluşturur. Bası semptomlarının derecesi açısından bazı hastalarda hiç belirti olmayabilir, bazı hastalarda ise çeşitli derecelerde sık tekrarlayan alt solunum yolu enfeksiyonları (bronşit, pnömoni vb), bazı hastalarda ise yutkunma güçlüğü, beslenememe ve kilo alma sorunları ortaya çıkar. Aberran sağ subklaviyan hastalarının çok büyük kısmında bu semptomlar hafiftir ve ömür boyu tolere edilebilir. Nitekim, çok küçük bir grupta bu semptomlar ağır olabilir, hayatın ilk yılında bu semptomlar ortaya çıkabileceği gibi 1 yaşından sonra hatta 4-5 yaştan veya kimi hastalarda da 10-15 yaşından sonra semptomlar ortaya çıkabilir. Hastalarda ameliyat kararını verdirecek en önemli kriter hastanın semptomunun yani şikayetlerinin olmasıdır. Bu sebeple çocuğunuzun eğer hiçbir semptomu veya şikayeti olmazsa ya da şikayetleri çok hafif ise ameliyata gerek yoktur. Hastalığın her tespit edildiği hastada ameliyat yapılması gerekir diye bir kural bulunmamaktadır. Yani semptomu olmayan hastayı ameliyat edip sonra da bir ömür boyu boşuna mı ameliyat ettirdik diye düşüneceğiniz bir durum olmayacağına emin olabilirsiniz. Doğum sonrası tetkikleriyle tarafımıza başvurursanız yardımcı olacağız.
Soru 10. Hocam benim 3 aylık bebeğim var 15 günlükken vsd tanısı konuldu 7 ml büyüklüğünde diyorlar ameliyat gerkiyormuş hocam ameliyat sorunsuz geçermi çok korkuttular beni çevremdeki kişiler nolursunuz yardımcı olun bana hocam ALLAH rızası için bebeğim yaşar mı hocam ameliyattan sonra cevabınız için şimdikten teşekkürler hocam saygılarımla...
Cevap: 10 yıllık vaka taramalarımızda 1 yaş altı geniş VSD'li hastalarda ameliyat sonrası ölüm oranımız % 1,4'tür. Yaptığımız çok daha kompleks ameliyatlar yanında nispeten riski daha düşük bir ameliyattır. Ama her açık kalp ameliyatında olduğu gibi bu ameliyatta da bir açık kalp ameliyatının taşıdığı riskler mevcuttur. Eğer bebeğin kilo alımı yeterli ise ve aylık eko kontrollerinde pulmoner hipertansiyonu ilerlemiyorsa (akciğer damar basıncı) 1 yaşına kadar beklenebilir. Ama biz bu tür hastalarda ilk 6 ay tercihen ilk 3 ay içerisinde vsd'yi kapatmayı tercih ediyoruz. VSD'nin erken kapatılması hastada kalıcı hipertansiyon gelişimini önlediği için ilerideki yaşamını da rahat geçirmesini sağlayacak en iyi tedbirdir. Bu bilgiler dışında çocuğun şu haliyle çok acil bir hayati tehlikesi bulunmamaktadır. Eğer bizimle irtibata geçerseniz yardımcı olacağız.
Soru 11. mrb lar oğlum şuan 27 aylık.17 günlükken akdeniz üniv.fallot+pulmoner artrezi+ad+vsd teşhisi kondu.1.şant ameliyatını 17 günlükken 2.şant ameliyatnı 15 aylıkken ege üniv.oldu.bu arda hiçbir sıkıntımız yok.herşey normal.15 gün önce anjiyo oldu.anjiyo sonucunda konsey kararına göre ameliyat olamaz takip edilsin kararı verildi.fakat bu arada şantlarınız kapanabilir çocuğu kaybedebilirsiniz dendi.kardiyoloğmuz bu düzeltici ameliyatının yapılabileceğini ve ancak oğlumuzun öyle kurtulacağını söyledi.yoksa işiniz çok zor dedi.bu ameliyatı yapan hastaneler arasında siyami erseği önerdi.cvb ınızı ve yorumlaınızı bekliyoruz.teşekkürler.
Cevap: Fallot+Pulmoner atrezi klasik Fallot Tetralojisinden farklı bir durum olup VSD+Pulmoner atrezi hastalık grubu içerisinde değerlendirilmektedir. Bu hastalarda pulmoner arterlere antegrad sağ ventrikülden çıkış atrezik yani kapalı olduğu için pulmoner arterler alternatif kaynaklardan kan alırlar. Bu kaynaklar arasında yenidoğan döneminde sıklıkla PDA (patent duktus arteriozus) varken yine yenidoğan döneminde veya ilerleyen dönemlerde MAPCA'lar bulunmaktadır. MAPCA'lar kollateral arterler olup sıklıkla desendan aortadan, aortanın dallarından veya bronşial arterler gibi aort dışı kaynaklardan çıkarlar. PDA veya MAPCA'lar yenidoğan dönemindeki kısa bir perioddan sonra sıklıkla yetersiz kalırlar. PDA'nın kapanması, MAPCA'larda olan stenozlar nedeniyle bu hastalar ilave kan kaynaklarına ihtiyaç duyarlar. Bu kan kaynakları sıklıkla cerrahi olarak oluşturulan şantlarla sağlanır. Şantlarla oluşturulan kan akımı ile pulmoner arterlerin gelişmesi sağlanarak normale yakın boyuta getirilmeye çalışılır. Pulmoner index denen ölçümlerle hastanın pulmoner arterlerinin yeterince gelişip gelişmediğine karar verilir. Pulmoner index'in çok düşük olduğu hastalarda tam düzeltme ameliyatı sonrası sağ kalp basıncı önündeki darlığa bağlı olarak çok yükselir ve sağ kalp yetmezliğine neden olur. Bu nedenle pulmoner arterlerin yeterince gelişmiş olması istenir. McGoon indexi denen indexin eskiden 2 ve üzerinde olması hastanın pulmoner arterlerinin yeterince gelişmiş olmasını göstermekteydi. Günümüzde ise pulmoner arter indexi 1,7-1,8 ve üzerinde olan hastalara veya lower lobe McGoon indexi 1,2 ve üzerinde olan hastalara da tam düzeltme ameliyatı uygulamaktayız.
Soru 12. KIZIM FALLOT AMEİYATI OLACAK RİSKİ NEDİR AMELİYAT SONRASI SORUNLARI TAMAMEM DÜZELECEKMİ
Cevap: Günümüzde Fallot ameliyatlarını %1 civarı ölüm oranları ile yapıyoruz. Genel olarak hastaların tekrar bir ameliyat ihtiyaçları olmuyor. Çok küçük bir hasta grubunda kalbin sağ tarafındaki kalp kapakçığında 10-12 yaşlar civarında yetersizlik olabiliyor. Günümüzde bu kalp kapakçığını ilk ameliyatta daha yüksek oranda koruduğumuz için artık bu oran da belirgin olarak azaldı. Fakat yine de bu kapağın (pulmoner) yetersizliği gelişen hasta grubunda eskiden beri ikinci bir ameliyatla biyolojik kapak yerleştirmekteydik. Nitekim gelişen teknolojiyle bu şekilde kalp kapakçığı yetersizliği gelişen hastalara ameliyatsız anjio katater ile biyolojik kapak yerleştirilebilmektedir.
Günümüzde Fallot ameliyatlarını 6 aydan itibaren yapmaktayız. Nitekim yoğun bakım süresinin kısalması ve ameliyat sonrası dönemin daha rahat olması gibi sebeplerle şikayeti olmayan hastalarda ortalama 1 yaşını ameliyat için tercih ediyoruz.
Soru 13. merhaba benim 3.5 aylık kızım var 1 aylıkken pm outlet 4mm vsd (70 mmhg gradiyet) asd/pfo tanısı kondu
sol boşluklar hafıf geniş önerilen sbe profilaksisi digoksin ,enapril,lasix başlandı 3 ay sonra kontrol denmişti
çocuk cerrahisi uzmanı bize 8 ayda ameliyat olması gerektiğini soyledi başkaca bir bilgi vermedi çok korkuyoruz lütfen bize daha detaylı bigi verebilirmisiniz.saygılarımla
şimdiki kılo:4.800
boy:63
ef-70 fs-36 interiyal septum 3-4mm asd7pfo
Cevap: 5 mm altı pm vsd'lerin %90'ı 1 yaşına kadar kendiliğinden kapanabilmekte olup hastanızdaki pm vsd'nin outlet yerleşimli olması kapanmasını güçleştirebilmektedir. Çocuğun kilo alımı yeterli ise ve yapılan kontrol ekokardiyografilerde pulmoner hipertansiyon düzeyinde ve konjestif kalp yetersizliğinde artma yoksa 1 yaşına kadar veya belki daha fazla da beklenebilir.
Hastanıza 3 aylık aralıklarla deneyimli ellerde ekokardiyografi yapılmasını öneriririm.
Soru 14. merhaba sayın hocam 2 ay bebk için banding ameliytı tavsiye ediliyor triküspit atrezisi mevcut siz önerıirmisiniz yoksa beklenmelimi asd ve vsd var yanı sıra ?
Cevap: Akciğer kan akımı normalden fazla olup saturasyonu yüksek tek ventriküllü triküspid atrezisi gibi hastalıklarda akciğer yatağını korumak için pulmoner banding 2-3 aylıkken yapılabilir. Uygundur...
Soru 15: Mrb,ben şuan 20 hft hamileyim ve bebekte down sendromu primum asd,inlet vsd,tek av kapak pulmoner stenoz var.(komplet avsd ve pulmoner stenoz)bebeğimi nezaman ameliyat Etmek uygundur?ameliyat sonrası yaşam şekli nasıldır başka ne gibi ek hastalıklar bizi bekler?ayrıca deneyim ve takiplerinize dayanarak ortalama bir yaşam süresi varmıdır?aydınlatırsanız çok mutlu olurum,teşekkürler
Cevap: Geçmiş olsun. Bu çocukları doğum sonrası ilk 6 ayda ameliyat ederek tam düzeltme yapmak en uygundur. Tercihen biz 4-5 aylıkken ameliyatlarını gerçekleştiriyoruz. Ameliyat sonrası komplikasyon gelişmeyen hastaların yaşam şekli ve süresi diğer down sendromlu çocuklarla aynıdır.
Doç.Dr.Buğra HARMANDAR
SORU:
Hocam oğlum henüz 5 günlük.Doğduğu hastahanede üfürüm teşhisiyle kardiyolağa gitmemiz istendi.
Raporunda;
Subaortik posterior malaligment gösteren 8*8 mm'lik vsd mevcut.Aorta %54 dekstropoze.Pulmoner arter ve dalları hipoplazik izleniyor.Pulmoner arterde kapakta başlayan turbülasyomn mevcut.APA 5 mm.Aort 3 kusplı.İAS fossa ovaliste anevrizmatik.Bu bölgede 2 mm'lik açıklık mevcut.Juguler çalışmada ince bir duktus akım alındı.Koarktasyon paterni alınmadı.Perikardiyal efüzyon yoktur.İnterventriküler septum hareketleri normaldir.Mitral valv prolapsusu yoktur.İdiyopatik hipertrofik stenoz görülmemiştir.
TANI : Subaortik malaligment geniş VSD (Fallot tetralojisi) ,Orta PS ,İAS anevrizması ,Küçük sekundum ASD ,Silent PDA
ÖNERİLER : AFN+/+ yazıyor.
Bunların anlamı nedir?Durum çokmu kötü?Tek bildiğim minik oğlumun kalbinde delik olduğu.Çok bir bilgi edinemedik belkide biz o an şoktan hiç birşey anlamadık.6 ay izleyeceğiz daha sonra ameliyat öngörebiliriz gibi birşeyler söyledi doktor hanım.Beslenmesi, uykusu normal, morarması yok.5 günde 150-200 gr kilo bile aldı.Bu durumun kendiliğinden iyileşme olasılığı hiç yokmudur?Neler yapmamız gerekir bu süreçte?Ameliyat sözkonusu olursa başarı şansı ve riski nedir.Açık kalp ameliyatıylamı olması gerekiyor operasyonu.Lütfen beni bilgilendirin yol gösterin..
CEVAP:
Çocuğunuzda Fallot Tetralojisi var. 6 ay - 1 yaş arasında açık kalp ameliyatı olması gerekiyor. Ameliyat başarısı hastanemizde çok yüksek olup ölüm oranı %1-2 civarındadır. Ameliyat dışında herhangi bir tedavisi yoktur. Ameliyat oluncaya kadar yalnızca enfeksiyondan korumanız yeterlidir.
Doç.Dr.Buğra HARMANDAR
SORU:
İyi günler kızımın 5 gunlukken mid muskuler vsd 2.7mm teşhis konuldu.2.ayda kontrole gelmemiz söylendi. 2.ay kontrolünde delik 2.5 mm ölçüldü. Suan 2.5 aylık kizimiz ve bir dahaki kontrol 6. ayinda. 3.5 aylikken uçak seyehati yapmasinda bir sakinca olur mu ayrica 2.5 mm olan vsd nin kapanma olasiligi nedir ameliyat gerektirecek bir durum olurmu? Kilo alimi cok normal ortalama günlük 25 gr kilo alıyor.
Cevaplariniz icin şimdiden teşekkür ederim
CEVAP:
2,7 mm'lik VSD çok küçük bir VSD olup pek önemi yoktur ve %90 ihtimalle 1 yaşına kadar kendiliğinden kapanır. Kapanmasa da genellikle ameliyat ihtiyacı olmaz.
Bu arada uçakla seyahatinde herhangi bir engel yoktur.
Doç.Dr.Buğra HARMANDAR
SORU:
HOCAM İYİ GÜNLER.
3.5 AYLIK BİR KIZIM VAR. DOKTORLARIMIZIN KOYMUŞ OLDUGU TEŞHİS SONUCU KALBİNDE 3 ADET DELİK OLDUĞU SÖYLENDİ. VE DOKTORUMUZUN VERDİĞİ İKİ ADET İLACIN YANINDA (İSİMLERİNİ AHTIRLAYAMIYORUM) DİĞOXİN VERDİ SABAH 2 DAMLA AKŞAM 1 DAMLA.
ALDIĞIMIZ RAPORDA;
KARDİYAL APEKS SOLA DOĞRU.
VENA KAVA İNFERİOR KOLUMMA VERTABRALİSİN SAĞINDA, İNEN AORT KOLUMMA VERTABLİSİN SOLUNDA (ATRİYAL SİTUS SOLİTUS)
İNTER ATRİYAL SEPTUMDA FOSSA OVALE BÖLGESİNDE 3 MM ÇAĞINDA SOLDAN SAGA SEKUNDUM ARTİYAL SEPTAL DEFEKT SAPTANDI.
İNTER VENTRÜKÜLER SEPTUMDA TRABÜKÜLER MÜSKÜLER BÖLGEDE 4 MM MODERATÖR BANT HİZASINDA VE HEMEN ONUN AŞAGISINDA APTİKAL BÖLGEDE 3-4 MM ÇAPINDA (TOPLAM 2 ADET) SOLDAN SAGA ŞANT GÖSTEREN VSD SAPTANDI. İNTERVENTRİKÜLER GRADİENT 84 MMHĞ ÖLÇÜLDÜ.
EN SON RAPOR KISMINDA İSE
MULTİPL VENTÜKÜLER SEPTAL DEFEK (TRABÜKÜLER VE APİKAL MUSKÜLER)
AST (SEKUDUM)
SAYIN HOCAM BEBEĞİMİZ İÇİN SIKINTI YARATACAK BİR DURUMMU.
KAPANMA İHTİMALİ VARMI . KAPANMA İSE;
ANJİYO İLE KAPATILABİLİR Mİ. AÇIK AMELİYAT OLMADAN.
TEŞEKKÜRLER.
CEVAP:
3 mm çaplı atriyal septal defektin pek önemi yoktur. Esasen 4 mm'lik ventriküler septal defekt de çok büyük bir defekt değildir ama sizin hastada 2 adet VSD olduğu için ikisi toplam 8 mm yapıyor ve hasta için önemli olabilir. Bu VSD'ler muskuler olduğu için kendiliğinden kapanma ihtimali var. Çocuğunuz için aylık eko kontrolü öneririm. Eğer 1 yaşına kadar kapanmaz ise veya büyüme gelişme kilo alma sorunu olursa kapatma yoluna gidebiliriz. Açık ameliyat olmadan da bazı durumlarda anjio ile kapatılabiliyor. Ama her hasta buna uygun olmuyor.
Geçmiş olsun.
Doç.Dr.Buğra HARMANDAR
SORU:
Hocam iyi günler. 11 yaşındaki oğlumu göğüs ağrısı şikayeti ile çocuk kardiyoloğuna götürdüğümde kalbinde delik ve kalp kapakçığında 2. derece yetmezlik olduğunu öğrendik. EKO sonucunu aynen yazıyorum.
Açıklama : İnteratriyal septumda 13 mm primum atrial septal defekt görülmüştür. Ventriküler seviyede geçiş görülmemiştir. 2. derece sol AV kapak yetmezliği görülmüştür. Hafif sağ AV kapak yetmezliği görülmüştür. (2,4 m/sn) Aort kapağı üç kasplıdır. Aorta ve pulmoner arter normaldir. Koroner arter çıkışları normal yerindedir. Arkus aorta solda yerleşimlidir. Aort koarktasyonu görülmemiştir.
SONUÇ : Parsiyel Atrioventrikluer Septal Defekt. 2. derece Sol AV Kapak Yetmezliği"
Çocuğun tedavisi için ameliyatın şart olduğu söylendi. İnternette yaptığım araştırmada bazı merkezlerde göğüs kafesi açılmadan koltuk altından girilmek sureti ile bu ameliyatın yapılabileceğini duyduk. sizin merkezinizde de bu şekilde ameliyat olma imkanı varmı. Lütfen yardımcı olursanız sevinirim.
CEVAP:
Hastanemizde Parsiyel Atrioventriküler Septal Defekt ameliyatlarını başarıyla gerçekleştirmekteyiz. Nitekim bu kalp ameliyatı bir açık kalp ameliyatı olup göğüs kafesi açılarak yapılmaktadır. Bu ameliyatın koltuk altından yapılmasının hastaya herhangi bir avantajı olmadığı gibi ameliyatın riskini arttırmaktadır. Bu nedenle koltuk altı girişimi bu ameliyat için önermiyoruz. Hastanemize başvurmanız halinde yardımcı olacağız.
Geçmiş olsun...
Doç.Dr.Buğra HARMANDAR
SORU:
doktor bey şu an 26+5 haftalık hamileyim ve bebeğimin kalbinde bi rahatsızlık olduğunu öğrendik bize direk ve kesin bir şekilde uğraşmayın yaşamaz yaşasada ameliyat falan gerekir yarım bir çocuk olur dediler ama ben bebeğimden vazgeçemem vazgeçmedimde ne gerekiyorsa yapmaya hazırız yaşaması için . dün Hacettepe ünüversite hastanesi kadın doğum bölümünden bir doktora gittim muayene ve kayıt için ama doktor bey bana bu hastanede bu ameliyatı yapmazlar daha kapsamlı kalp damar hastanelerine git tedavisi olur uzmanı daha iyi bilir ama şansı var dedi bu konu ile bilgi istiyorum raporlar ekte umarım vakit ayırır incelersiniz gel derseniz gelirim ben amasyada yaşıyorum burada çok kapsamlı hastane yada bu konuda tam anlamında uzman doktor yok ankara ve samsunda doktorlara gittim ama hepsi ameliyat olur ama bizde aldır dediler hocam internetten biraz araştırdım bu tarz ameliyatlar varmış lütfen bana geri dönüş sağlayın ... teşekkürler
CEVAP:
Geçmiş olsun. Çocuğu aldırmaya gerek yok. Biz esasen hiçbir doğumsal kalp Hastalığı'nda bebeğin alınmasını önermiyoruz (çünkü riski yüksek olsa da tüm doğumsal kalp hastalıkları olan çocuklar ameliyatlarla yaşatılabiliyor), nitekim sizin çocuğunuz da mevcut kalp Hastalığı'na yönelik ameliyatlarla yaşatılabilir. Kalbinin sağ tarafı gelişmemiş ve sağ akciğere giden damarının dar olduğu bir hastalığa sahip. Doğumdan sonra ilk 15 gün içerisinde Şant adını verdiğimiz bir ameliyatın yapılması gerekiyor. Çocuk doğar doğmaz Prostavazin isimli bir ilaç başlanacak ve 15-20 gün içerisinde ameliyat olacak. Bu ameliyatta akciğere kan vermek için suni bir damar takılacak kalbine. Bu ameliyatın hayati riski %20-30 civarındadır. Bu ameliyatı sorunsuz atlatan ve iyi gelişen hastalara 1 yaş civarında Glenn Şant ismini verdiğimiz bir ameliyat yapıyoruz. Bu ameliyatla suni damardan kurtuluyor ve büyüyüp gelişmeye başlıyor. Bu ameliyatın hayati riski %5-10 civarındadır. Iyi büyüyüp gelişen hastalara 4-5 yaş civarında Fontan isimli üçüncü ve son ameliyatı yapıyoruz. Bu ameliyatın riski %10-20 civarındadır. Bu ameliyatı sorunsuz atlatan hastalar hemen hemen normal bir insana yakın oksijen düzeylerine kavuşuyor ve dıştan bakıldığında normal bir insandan pek farklı olmuyorlar. Bu şekilde ameliyatlarını tamamladığımız 18 yaşında hastalarımız var. Yani çocuğunuz ümitsiz değil. Sadece riskli ameliyatlara aday. Çocuk doğar doğmaz ekosunu yaptırıp bahsettiğim ilacı başlatıp beni ararsanız yardımcı olacağım.
Geçmiş olsun,
Doç.Dr.Buğra Harmandar
SORU:
Merhaba doktor bey, 2 aylık kızımda doğduğundan itibaren üfürüm var.doktora götürdük PM QUTLET VSD 5MM, SEKUNDUM ASD 4-5 MM,TRİKÜSPİT KAPAK POŞ YAPIYOR. HAFİF PULMONER HİPERTANSİYON,KİLO TAKİBİ ,DİGOKSİN BAŞLANDI. Tanısı kondu. kilosu: 4600gr 2 ay sonraya kontrol verildi yaşadığım yer DÜZCE küçük bir yer daha büyük bir sağlık kuruluşuna gitmeliyim, Doktor deliklerin kapanıp kapanmaması konusunda net bir şey söylemedi.bu konu hakkında bilgi verirseniz sevinirim
CEVAP:
Perimembranöz outlet VSD'ler genellikle kendiliğinden kapanmaz ama bazen kapanabiliyor da. 5 mm VSD büyüklüğü açısından sınırdır. 5 mm'nin üstü geniş VSD olup genellikle kapanmayacağı düşünülür, 5 mm altı küçük VSD'dir ve kapanma ihtimali yüksektir. Eğer 2 ay sonra çocuğunuzun kilo alımı yeterli değil ise ve büyüme gelişme geriliği oluyorsa, VSD de kapanmamış ise o zaman ameliyatla kapatabiliriz. Cerrahisinde hayati risk günümüzde %1-5 düzeylerine kadar düşmüştür. İrtibat telefonlarım aşağıda. 2 ay sonra görüşelim.
Geçmiş olsun,
Doç.Dr.Buğra HARMANDAR
Konu : anne karnında kalpte delik
Mesaj: Merhaba, Şuan tam 22 haftalık hamileyim. Dün gittiğim detaylı ultrosonda bebeğimin kalbinde 3,2 ve 1,6mm ölçülerinde 2 delik olduğunu öğrendim. Doktorum bunlardan küçük olanın doğuma kadar kapanacağını diğerininde 1 yaşına kadar kapanabileceğini söyledi. Fakat ben endişeleniyorum. Doğumda bebeğime bir şey olur mu bu down sendromuna işaret midir veya doğum sonrasında hayatını etkileyecek bir soruna dönüşür mü? Raporu kısaca yazmak istiyorum; SAT ile uyumlu 22 haftalık gebelik. Yapılan detaylı ultrason incelemesinde fetusta kalpte 3,2 ve 1,6 mm lik musküler VSD izlendi. Bunun dışında yapısal anomali görülmedi. Böylece doğumdan sonra ortaya çıkabilecek anomalilerin %70-80''i dışlanmıştır. Kalbin dört odacığında, aorta ve pulmoner arter çıkışları ve üç damar kesiti normal olarak değerlendirilmiştir. ARSA yok. Bilgi verirseniz sevinirim. Hayırlı günler.
Cevap:
Geçmiş olsun,
Korkacak bir durum yok. Anne karnında VSD tespit edilmesi ile down sendromunun bir ilişkisi yoktur. Down olanlarda VSD daha fazla olabiliyor ama her vsd olan down sendromu olmuyor. Bebeğinizin vsd 'leri çok küçük. Muhtemelen her ikisi de doğuma kadar kapanabilir. Kapanmasa bile bu kadar küçük delikler genellikle 1 yaşına kadar %90 oranında kapanırlar. 1 yaşına kadar kapanmasa da 4-5 yaşına kadar herhangi bir müdahaleye gerek yoktur. Zaten 4-5 yaşına kadar da kapanma ihtimali devam etmektedir.
Soru:
iyi günler, 2011 yılında kızım 15 aylıkken fallot tetralojisi tanısı konularak ameliyat edildi. Düzenli kontrolları yapıldı. 8 nisan 2015 tarihinde kontrol yapılırken trisküpirit kapak normal renkli doppler ile hafif yetersizlik saptandı yetersizlik hızı 3 m/s pulmoner grandient:23mmHg. pulmoner arter dalarından itibaren 1,8 m/s akım hızında yetersizlik izlendi., doktor tekrar bir ameliyat gerekebilir dedi kalbinin sol tarafında büyüme olduğunu söyledi, sizce bu durumda hayati tehlikesi var mı?
Cevap:
Fallot nedeniyle ameliyat edilen hastaların bir kısmında yapılan cerrahi tekniğe bağlı olarak kalbin sağ tarafındaki pulmoner kapakta yetersizlik meydana gelebiliyor. Bu tür hastalarda kardiyak MR yaparak sağ ventrikül end diastolik hacmini ölçüyoruz. Eğer bu hacim 150 ml'nin üzerinde ise, sağ kalpte kasılma kusuru meydana geliyorsa veya hastada şikayetler başlamışsa yeni bir ameliyat yaparak sağ kalp çıkım yoluna pulmoner kapak yerleştiriyoruz.
Geçmiş olsun,
Soru:
Merhabalar benim 3,5aylik kizima dogumdan once pulmoner atrezi+vsd teshisi kondu. 30 gunlukken sant ameliyati yapildi pda kapatildi. Ogrenmek istedigim su ki internette bize aciklanan tdaviye benzer bi yaziya hic denk gelmedim. Pulmuoner arterinin olmadigi eger akciger damarlari gelisirse 1 yas civarinda duzeltme ameliyati olabilecegi soylendi. Bu duzeltme ameliyatinda hayvandan alinan damar takilacak ve bu damar kuculdukce/yiprandikca ortalama 5 yilda bir yenilenecek, kizim omru boyunca ameliyat olacak. Bu hastaligin baska bir tedavi sekli var midir yardimci olursaniz sevinirim.
Cevap:
Geçmiş olsun,
1 yaş civarında akciğer damarları uygun hale geldiğinde hayvandan alınan kapaklı kondüit ile tam düzeltme yapıyoruz. Ardından bu damar 3-5 yıl içerisinde ya küçük kalıyor ya da yıpranıyor, o yüzden değiştirmek gerekiyor. Bu şekilde çocuğunuz ömür boyu ameliyat olmayacak. Yerleştirilen yeterli genişlikte bir kapaklı kondüitten sonra artık anjio katater ile bu damarın içerisine girip ameliyatsız kapak yerleştirmek ve daralmış damara balon yapma imkanı olabiliyor. Bu şekilde yaptığımız birkaç hastamız var. Bu konuda teknoloji çok hızlı ilerliyor. İnşallah ileride 1-2 kere bu kapaklı kondüiti değiştirdikten sonra kalan iş anjio katater ile halledilebilecek gibi görünüyor.
Soru:
Hayirli gunler 19aylik oglumdj 12mm delik tespit edildi kilo 15.5 asd nasil oluyor kapanirmi.
Cevap:
Küçük asd'ler 2 yaşına kadar kapanabiliyor. Ama sizin çocuğun ASD'si büyük olduğu için kapanması pek beklenmez. 4-5 yaşına kadar herhangi birşey yapmaya gerek yok. 4-5 yaşında değerlendiririz. ASD'si önemli ise ve o zaman anjio katater ile kapanmaya uygun ise ameliyatsız kapatılır. Uygun değilse ameliyat gerekebilir. Ameliyatı diğer kalp ameliyatları içerisinde en başarılı olan ve ölüm oranı hemen hemen %0 olan bir ameliyattır. Olabildiğince estetik yapma imkanı da vardır. Geçmiş olsun, görüşmek üzere,"
Doç.Dr.Buğra HARMANDAR
Soru:
mrb hocam 35 günlük oğlum 6 günlükken şant amalıyatı oldu amalıyat basarılı geçtiği söyleniyor ama oğlum hala solunum makınesınden ayrılamadı 6 defa denemelerıne rağmen doktoru boğazından bi delık açıp tüp yerlestırmeyı planlıyor ancak o zaman ayrılabilir dıyor daha öncede sıze yazmıstım sonuçları tga ps demiştiniz hocam yoğunbakımdan çıkınca bu tüp ne zamana kadar kalır bilgilendirirseniz sevinirim teşekkürler
Cevap:
Çocuk uzun süre solunum cihazından ayrılamazsa trakeostomi denilen boğazına delik açma işlemi uygulanıyor. Bu şekilde solunum cihazından ayrılabilirse bazen birkaç hafta bazen de birkaç ay sonra boğazındaki kanül çıkarılıyor. Çocuğunuza yapılan ameliyat geçici bir ameliyattır. Esas ameliyatı 1 yaşları civarında yapılması gerekiyor. Geçmiş olsun,"
Doç.Dr.Buğra HARMANDAR
Soru:
Merhaba Hocam
Eşim 20 haftalık gebe.19+5 günlükken Perinatoloji Uzmanı tarafından yapılan detaylı ultrason muayenesinde Situs inversus torakalis,ventriküler septal defekt,sol ventrikül hipoplastik,pulmoner atrezi ve sağ persistan umbilikal ven tespit edildi.
Kompleks bir kalp anormalliği olduğu söylendi.Anne karnında ölebileceği doğsa bile hemen ameliyat edilmesi gerektiği ve ameliyat edilse bile yaşamasının zor olduğu söylendi.Terminasyon seçeneğini de düşünmemiz söylendi.
Sizce ne yapmalıyız?
Cevap:
Anne karnında çocuğun kan dolaşımı ve oksijenlenmesi göbek kordonu ile desteklendiği için çok büyük ihtimalle ölüm olmaz. Doğum sonrası ilk 15 gün içerisinde ameliyat edilerek düzeltilmesi gerekir. Ameliyatlar yüksek risklidir ama bu ameliyatları yaptığımız yaşayan pekçok hastamız vardır. Geçmiş olsun."
Doç.Dr.Buğra HARMANDAR
Soru:
merhaba buğra bey,29 haftalık hamileyim siymi ersek hastanesinde 6 martta muay oldum.teşhiste sol artriyum sol ventrikul ileri derecede hipoplazik.ve sağ kalp boşlukları genişdr bey yaşamaz dedi ameliyat bile olsa çok zor dedi..ikinci bir çocukta aynı hastalık olur mu aklıma şimdi bunlar geliyor sürekli...
Cevap:
İkinci çocukta olma ihtimali diğer insanlarla aynıdır. Hipoplastik sol kalp ameliyatları yüksek riskli ameliyatlar olup ülkemizde biz dahil pekçok merkezde yapılmaktadır."
Doç.Dr.Buğra HARMANDAR
Soru:
hocam 1 yaşında kızımın eko sonucu aşağıda.sorun tam olarak nedir ve geçer mi
TRANSTORASIK EKOKARDIYOGRAFI, < 4 YAŞ ÇOCUK
KALP HIZI:162/DK
PFO(INFERIYOR YERLESIMLI)
VENTRIKUL BOYUT FONKSIYONLARI NORMAL
PDA, AORT KOARKTASYONU SAPTANMADI
6 AY SONRA POLIKLINIK KONTROLU
8 AY SONRA EKOKARDIYOGRAFI KONTROLU
PFO(INFERIYOR YERLESIMLI)PFO(INFERIYOR YERLESIMLI)
Cevap:
Kalbinin iki kulakçığı arasında küçük bir delik var. 4-5 yaşına kadar kendiliğinden kapanma ihtimali yüksek. Kapanmasa bile genellikle ömür boyu sorun yaratmaz."
Soru:
Hocam merhaba, doğum kontrolleri sırasında yapılan detaylı ultrasonlarda ora genişlikte olan (6,5-7 mm) vsd tespit edilebilirmi. ayrıca bu durum tespit edilmiş bebekler için normal doğumun hayati risk oluşturduğu, sezeryanla doğum yapılması gerekliliği ne kadar dır?
Cevap:
Fetal eko ile tespit edilebilir. VSD bulunan bebeklerin normal doğumla doğmasında herhangi bir sakınca yoktur."
Soru:
Merhaba,
Şuan tam 22 haftalık hamileyim. Dün gittiğim detaylı ultrosonda bebeğimin kalbinde 3,2 ve 1,6mm ölçülerinde 2 delik olduğunu öğrendim. Doktorum bunlardan küçük olanın doğuma kadar kapanacağını diğerininde 1 yaşına kadar kapanabileceğini söyledi. Fakat ben endişeleniyorum. Doğumda bebeğime bir şey olur mu bu down sendromuna işaret midir veya doğum sonrasında hayatını etkileyecek bir soruna dönüşür mü?
Raporu kısaca yazmak istiyorum;
SAT ile uyumlu 22 haftalık gebelik. Yapılan detaylı ultrason incelemesinde fetusta kalpte 3,2 ve 1,6 mm lik musküler VSD izlendi. Bunun dışında yapısal anomali görülmedi. Böylece doğumdan sonra ortaya çıkabilecek anomalilerin %70-80'i dışlanmıştır. Kalbin dört odacığında, aorta ve pulmoner arter çıkışları ve üç damar kesiti normal olarak değerlendirilmiştir. ARSA yok.
Bilgi verirseniz sevinirim. Hayırlı günler.
Cevap:
Geçmiş olsun, Korkacak bir durum yok. Anne karnında VSD tespit edilmesi ile down sendromunun bir ilişkisi yoktur. Down olanlarda VSD daha fazla olabiliyor ama her vsd olan down sendromu olmuyor. Bebeğinizin vsd 'leri çok küçük. Muhtemelen her ikisi de doğuma kadar kapanabilir. Kapanmasa bile bu kadar küçük delikler genellikle 1 yaşına kadar %90 oranında kapanırlar. 1 yaşına kadar kapanmasa da 4-5 yaşına kadar herhangi bir müdahaleye gerek yoktur. Zaten 4-5 yaşına kadar da kapanma ihtimali devam etmektedir. "
Soru:
Merhaba hocam benim 3.5 aylık oğlum var üfürüm sonucu biküspit aort aort stenoz varmış kalbinde rapor şu şekilde:kalp boşlukları normal genişlikte ve dengeli boşluktadır İyi ve İdari intakttır. Sağ ventrikül çıkım yolu açıktır. Aort kapak biküspittir. Aort kapakta trübülans görülmüştür, CD doppler ortalama 27mmHg,maksimum45mmHg sistolik gardiyan elde edilmiştir. Aort kapakta minimal, triküspit kapakta minimal yetmezlik belirlenmiştir. Suprasternal çalışmada duktus, koarktrasyon görülmemiştir. Sol ventrikül fonksiyonu normaldir. ( çıkan aort :13.4mm) bu ikinci kontrol ilk kontrolde darlık 36_40 MmHg idi hocam gebelikte gayet sağlıklı dendi şuanda gelişimi gayet iyi deniliyor. bu durum oğlumun yaşam kalitesini olumsuz etkiler mi yardımcı olursanız sevinirim teşekkürler ederim.
Cevap:
Biküspid aort kapaklarda darlık, yetersizlik ve bunlara bağlı olarak asendan aortta genişleme olabiliyor. Sizin çocukta aort kapakta hafif darlık, hafif yetersizlik mevcut. Asendan aortta genişleme yok. Acil müdahale gerekmiyor. 6 aylık aralıklarla eko takibi önerilir."
Doç.Dr.Buğra HARMANDAR
Soru:
merhaba hocam kızım 7 yaşında doğuştan pda üfürüm var diyorlar doğduğunda 0,7ml şuan 03ml acaba zamanla kapanabilirmi? çünkü kızımda çok şükür hiç bir hareket kısıtlanması yok yaşıtlarından önde gidiyor hangi doktara gittiysek anjiyo olup katater adında bir telle kapatarız diyor biz operasyon olmasını istemiyoruz. sizce ileride bir sıkıntı çıkarmı? görüşlernizi merak ediyorum vakit ayırdığınız için teşekkür ederim
Cevap:
Anjio katater ile kapatılabileceği doğru. Eğer sol kalpte yüklenme bulguları varsa kapatılması uygundur."
Soru:
merhaba doktar bey kızım 1 aylıkken 4mm ads izlendi 6 aylıkken başka bir doktorda 5 mm olarak izlendi sizce kapanma olasılığı yüksekmi yoksa büyüme yapabilirmi. herhangi ciddi bir rahatsızlık verebilirmi
Cevap:
4-5 mm asd küçük bir asd'dir ve ciddi bir rahatsızlık vermez. Genellikle kendiliğinden kapanır. Eğer 4-5 yaşına kadar kapanmaz ise gerekli olduğu durumlarda anjio katater ile ameliyatsız veya cerrahi olarak başarılı bir şekilde kapatılabilmektedir."
Soru:
merhaba hocam 34 haftalık gebeyım doguma az kaldı perınatolojı doktorumuz 20. haftalarda aort koartasyonu oldugunu söyledı doguma çocuk kardilojısının girmesı gerekıyomuş orasını anladım ama böyle vakalarda sonuç ne oluyo başarı oranı ameliyat başarılı geçse her şey normale döner mi bizi ne beklıo .dogum yaklaştıkça bu konuda korkularım artıo bilgilendirirsnız sevinirim teşekkurler.
Cevap:
Basit aort koarktasyonlarının cerrahi sonuçları çok iyi olup ölüm oranları %1 düzeylerindedir. Çocuğunuz doğduktan sonra yapılacak eko ile değerlendirilip ameliyat olup olmayacağına karar verilecektir. Geçmiş olsun,"
Soru:
Hocam merhaba.
Kizimiz 13 gunluj..bugun eko cektirdik..sonucta ince pda ve pfo yaziyor..ve 1 yasinda konrol yaziyor...hocam korkacak bir durum varmi..ilerdeki karsilasacaklarimiz hakkinda bilgi verebilirmisiniz..ve ayrica bu 1 yil icerisinde kacinmamaiz gereken durum hareket varmi..tesekkurler..
Cevap:
Çok ağır bir durum değildir. İnce pda ve pfo çocukta kalp yetersizliği veya büyüme gelişme geriliği yapmaz. Eğer 1 yaşına kadar pda kapanmazsa ameliyatsız anjio katater ile başarıyla kapatılabilmektedir. Çocuğun enfeksiyondan korunması dışında alınabilecek ilave bir tedbir yoktur."
Soru:
Oğlum 3,5 yasonra Asd denildi 18mm cerrahi şartımız şemsiye yöntemi olurmu yada bi ümit kapanırmı size nasıl ulaşabiliriz randevu için
Cevap:
18 mm ASD'nin kendiliğinden kapanması pek beklenmez. Şemsiye dediğiniz device ile kapama belki olabilir ama uygun olup olmadığına kardiyologlar tarafından karar verilmesi gerekiyor. Bu işlem için de sıklıkla TEE denilen eko yapılıyor. Eğer device ile kapamaya uygun değilse cerrahi riski düşüktür ve başarıyla yapılmaktadır. Ayrıntılı eko raporu varsa [email protected] mail adresime gönderirseniz değerlendireyim."
Soru:
mrb hocam benim 4 aylık oğlum 6 günlükken bt şant amalıyatı oldu şuan durumu iyi dosyasında pulmoner artezi asd vsd bat yazıyor vsd 14mm asd 7mm hocam benim oğlumun daha kaç amalıyat olması gerekıyor ve hangi amalıyatları olucak ve amalıyatların riski ne kadardır bizleri aydınlatırsanız seviniriz hocam çok teşekkürler
Cevap:
Eğer pulmoner arter gelişimi iyi olursa 6 ay - 1 yaş arası tam düzeltme yapılabilir. Eğer yeterli olmazsa beklenebilir veya santral bir şant ya da sano şantı gerekebilir. Tam düzeltme yapıldıktan sonra yerleştirilecek olan biyolojik kapaklı kondüitlerin 3-5 yıl ömrü var. 3-5 yıl sonra değiştirip daha büyüğünü takmak gerekiyor. Ama belirli bir çapa ulaşıldıktan sonra ameliyatsız içerisine anjio katater ile balon yapmak ve yeni bir kapak yerleştirmek mümkündür. Yapılacak ameliyatların tamamı riskli ve zor ameliyatlardır. Geçmiş olsun."
Soru:
Öncelikle merhaba benim kızım 10 yaşında transkateter asd kapatılması yapıldı ama raporunda septum orta kısımda cihaz yerinde izleniyor cihazın AV kapaklar tarafındaki ucunda soldan sağa şantlı çok ince geçiş izlendi diyor bununla ilgili bilgilendirirseniz çok sevinirim Teşekkürler 27.05.2015 oldu operasyonu kızım hocam normalde 1 ay sonra gidecekdik ama doktor 10 gün sonra dedi bu bir sorun olurmu acaba bilgilendirirseniz sevinirim
Cevap:
Device kenarından çok ince bir geçiş olduğu yazıyor. Bu durum çok büyük bir ihtimalle birkaç ay içerisinde o bölgede geliişecek endotel oluşumu ile kapanacaktır. 10 gün sonra bakılacak eko sonrası aylık eko kontrolü uygun bence. Geçmiş olsun,"
Soru:
Merhaba hocam.oğluma 3 günlükken vsd teşhisi konuldu şuan 3 aylik. Size 3.aydaki raporunu yazıyorum
Vsd (perimembranöz,hafif malalignment,5mm,lr grad 30mmHG) vsd apikal muskuler ASD(sekundum 3-4mm)Pulmoner stenoz(valvüler ve infindibular maks grad 65mmhg) ve şuan 6 kilo gelişimde bir sorun yok. Doktorumuz %90 1 yaşında ameliyat yapacagiz dedi. Siz ne düşünüyorsunuz ameliyat şartmi yaptirmalimiyiz kapanır mı kendiliginden çok korkuyorum ve üzülüyorum.
Cevap:
Evet, şu anda acil ameliyata gerek yok. Önemli pulmoner stenoz olması VSD'nin akciğere getireceği volüm yükünü azaltıyor. Büyüme gelişmesi iyi olduğu sürece 1 yaşına kadar beklenebilir. Yalnız apikal VSD cerrahi olarak değil de perventriküler device ile de kapatılabilir belki ama pulmoner stenoz balon anjioplasti ile tam açılır mı bilemiyorum çünkü infundibuler darlığı da var. 3 ayda bir eko takibi uygun. 1 yaş civarı karar verilebilir. Geçmiş olsun,"
Doç.Dr.Buğra HARMANDAR
Soru:
Hocam merhaba, 2,5 aylık kızıma atriyal septal defekt teşisi kondu. Eko raporu aşağıdaki gibi, ne yapmamız gerekiyor, kendiliğinden kapanır mı yoksa müdahale mi gerekiyor? yorum yapabilirseniz çok sevinirim. Sağ kalp boşlukları normalden ve hafif sol kalp boşluğundan geniş, kalp kontraksiyonları normal sınırlar içinde. kardiyak apeks sola doğru. vena kava inferiyör kolumna vertebralisin sağında, inen aort kolumna vertebralisin solunda (atriyal situs solitus) Atriyumların ventrikülerle ve ventriküllerin büyük damarlarla olan ilişkisi normaldeki gibi. sistemik ve pulmoner venlerde dönüş anomalisi saptanmadı. İnter atriyal septumda sekundum 2 D çapı 6,4 mm, renkli akım çapı 7 mm genişliğinde soldan sağa laminer vasıfta şantlı defekt görüldü. Mitral - triküspit kapaklar normal, renkli dopler ile belirgin bir yetersizlik saptanmadı. İnter ventriküler septumda defekt görülmedi. Aort ve pulmoner arterin ilişkisi normaldeki gibi. aort kapağı triküspit, renkli veHPRF dopler ile aort akımı normal , yetersizlik saptanmadı. Pulmoner arter hafif geniş. Her iki pulmoner arter dalları konfluent. renkli ve HPRF dopler incelemede pulmoner arter akımı hafif türbülan ve akım hızı artmış bulundu. APA akım hızı: 1,56 m/s . Suprasternal incelemede aort arkusu solda. Aort koarktasyonu ve patent duktus arteriyosus görülmedi.Perikardiyal effüzyon tespit edilmedi.
Cevap:
Bu çok geniş bir ASD değil. Orta genişlikte. 2 yaşına kadar kapanma ihtimali var. Ama sınırda olduğu için kesin kapanır diyemeyiz, açık da kalabilir. Eğer küçük olsaydı kesin kapanır veya büyük olsaydı kesin kapanmaz diyebilirdik. Eğer kapanmaz ise 3 yaşından sonra ameliyatsız anjio katater ile device yerleştirilerek kapatma denenebilir. Eğer başarılı olmaz ve yine de kapatma ihtiyacı olursa 5 yaş civarı cerrahi olarak kapatılabilir.
Merhaba Hocam, 4,5 yaşında oğlumu kalpte üfleme teşhisiyle çocuk Kardiyoloji ye götürdük. Sonuç ta ASD (sekundum; multifenestre) sağ kalp boşluklarında genişleme yazıyor. Kalın puntolarla LVDd 3.6 cm RVDd 2.5 cm Atriyal septum: sekundum bölgede 2 çapı 15.4 mm, renkli akım çapı 16.1 mm olan soldan sağa şantlı defekt saptandı.ilave olarak bu defektin3mm uzağında ikinci bir defekt görüldü. AV kapak rimi 6.5 mm IVC rimi deficient. Kalp boşlukları : Sağ kalp boşlukları geniş yazıyor. Doktorlar heyet e sunup kapalı mı açık mı operasyon için karar alacak. Başarı oranı, kapalı mı açık mı olmalı sizce düşüncelerinizi öğrenebilir miyim Teşekkürler
Cevap:
Çocuğunuzun eko değerlerini inceledim. Kapalı olarak katater ile device yerleştirilmeye bence pek uygun görünmüyor. Ama çok deneyimli kardiyologlar uygun görürlerse device yerleştirebilirler belki. Eğer kapalı olarak device yerleştirmeye uygun çıkmazsa ameliyat sonuçları çok iyidir. Biz de bu tür hastalarımıza mini göğüs kesisi ile ve estetik cilt kapama ile olabildiğince minimal invazif ameliyatlar yapıyoruz.
Başvurmanız halinde yardımcı olabiliriz.
Geçmiş olsun,
Soru:
iyi günler hocam BENİM 36 GÜNLÜK BİR OĞLUM VAR KOMPLET ATRİYOVENTRİKÜLERSEPTALDEFEKT BALANCED VENRİKÜLER SEPTAL DEFEKT İNLET ATRİYALSEPTAL DEFEKT PRİMUN+SEKENDUM TEK AV KAPAK YAPISI AV KAPAKYAPISI AV KAPAK YETERSİZLİĞİ HAFAF ORTA PULMONER HİPERTANSİYON HAFİF TANI KONULDU CAPRİL VE LATEKS İLACA BAŞLANDI DOĞUM KİLOSU 3090 ŞUAN 3500GR KİLO ALMASI İÇİNDE TAKVİYE MAMA TARZI ECZANEDEN İLAC ALIYORUZ BU AMELYAT TEK SEFERDE GERCEKLEŞİRMİ YOKSA DAHA FAZLA BİR AMELYAT SÜRECİ OLURMU ŞİMDİDENTEŞEKKÜRLER
Cevap:
Balanced komplet atrioventriküler av septal defekt tanılı hastalarda modifiye tek yama ve çift yama teknikleri ile hastaların yapısal özelliklerini dikkate alarak tam cerrahi düzeltme yapıyoruz. Normal şartlar altında tek seferde tam düzeltme yapıyoruz. Nadiren ileri dönemlerde kalp kapaklarında yetersizlik oluşursa bu yetersizliklere yönelik tedaviler de yapıyoruz. Bu ameliyatların günümüzde başarı oranı oldukça yükselmiş ve cerrahi ölüm oranları %5 civarlarına düşmüş bulunmaktadır. Ameliyat zamanlaması olarak ortalama 4,5 - 5 ay aralığını seçiyoruz. Ama hastaların durumuna göre bu yaş daha erken olabiliyor (kilo alamama, büyüme gelişme geriliği, kalp yetersizliği vb durumlarda) veya bazen 6 aylığa kadar uzayabiliyor.
Geçmiş olsun,
Soru:
VSD'li TGA 'larda ameliyat ne zaman yapılmalıdır ?
Cevap:
VSD ile birlikte olan TGA 'larda doğumdan sonra ne zaman ameliyat yapılacağına VSD'nin büyüklüğüne bakılarak karar verilir. Eğer geniş bir VSD ise ameliyat için hasta doğum sonrası birkaç ayı hatta bazen daha fazla bekleyebilir. Çünkü geniş VSD nedeniyle kalbin sol tarafı korunur ve ameliyat şansını kaybetmez. VSD orta genişlikteyse genellikle ilk 1 ay içerisinde ameliyat yapılabilir. Ama eğer vsd küçük ise o zaman genellikle doğum sonrası ilk 15 gün içerisinde ameliyatını yapmak gerekiyor.
Geçmiş olsun,
Soru:
Sayın hocam iyi akşamlar,ben 21 haftalık hamileyim,bebeğimde ekte gönderdiğim teşhisle kalp rahatsızlığı çıktı,operasyon gerektiği anlatıldı,Eylülde tekrar kontrole gidip baktırdığımda doğar akciğer oluşumuna göre doğar doğmaz mı yoksa daha sonra mı olacağı belli olacak..şimdi operasyon yapacak doktor arayışına girince adınızı ve bu tür ameliyatları sıkça yaptığınızı duydum...böyle bir tabloda risk durumu nelerdir,tamamen sağlığına kavuşma olasılığı var mıdır? kafamızda ebeveyn olarak türlü sorular var..bizi aydınlatabilirseniz sevinirim,şimdiden vakit ayırdığınız için çok teşekkür ederim....saygılarımla
Cevap:
Geçmiş olsun,
Çocuğunuzun eko raporunu inceledim. Triküspid atrezisi yani kalbin sağ tarafındaki kapakçığın gelişmeme sorunu var. Bu sorun nedeniyle kalbin sağ tarafı da normale göre küçük durumda. Böyle bir hastada doğum sonrası kalbin sağ tarafını tekrar büyütüp normal haline getirme imkanı olmadığı için hastanın yaşına ve durumuna göre kalbin sağ tarafını bypass ederek kirli ve temiz kanı birbirinden ayıracak bazı ameliyatlar yaparak hastayı yaşatabiliyoruz. Öncelikle bu triküspid atrezisinin 3 formu olduğunu söylemeliyim. Akciğere giden damarda ciddi daralma veya tıkanıklık olan tiplerinde (sizin ekoda böyle bir durum belirtilmemiş) doğumdan sonraki ilk 1 hafta 15 gün içerisinde akciğer damarına kan verecek şekilde şant ismini verdiğimiz bir suni damar yerleştirme ameliyatı yapıyoruz. Akciğere giden damarda genişleme olanlara ise doğumda herhangi bir müdahale yapmayıp 2 ay civarı pulmoner banding ismini verdiğimiz bir daraltma işlemi yapıyoruz. Bu iki grup arasında kalıp akciğere giden damarda hafif darlık olan bazı hastalara ise erken dönemde herhangi bir ameliyat gerekmeyebiliyor. Daha sonra bu çocuklar 6 ay - 1 yaş arasına geldiğinde kalbin sağ tarafını bypass ederek vücudun üst kısmından gelen kirli kanı temiz kana karışmadan ayıracak ilk ameliyat olan Glenn şant ismini verdiğimiz bir ameliyat yapıyoruz. Bu ameliyatla çocuklar %90 ve üzeri bir oksijen satürasyonuna kavuşuyor ve diğer çocuklara yakın bir büyüme gelişme gösterebiliyorlar. Bu ameliyatla 4-5 yaşına kadar devam ettikten sonra üçüncü ve son ameliyatı yapıyoruz. Fontan ismini verdiğimiz son ameliyatta vücudun alt kısmından gelen kirli kanı kalbe dökülmeden ayırıp akciğer damarına birleştiriyoruz. Bu son ameliyat ile hastalar yine %90 ve üzeri bir satürasyonla normal insanlara yakın oksijen oranları ile hayatlarına devam edebiliyorlar. Yenidoğan döneminde yapılanlar daha riskli olmak üzere tüm ameliyatlar riskli ve ağır kalp ameliyatlarıdır. Daha önceden bu ameliyatlardan çok fazla miktarda yaptığımız için tecrübemiz fazla. Eğer, isterseniz doğum sonrası yapılacak ilk eko ile çocuğu değerlendirip ameliyat programına alabiliriz.
Prof.Dr.Buğra HARMANDAR
Yorumlar- Yorum Yaz
çamaşır makinesi ses çıkarması topuz modelleri kapalı huawei hoparlör cızırtı hususi otomobil fiat doblo kurbağalıdere parkı ecele sitem melih gokcek jelibon 9 sınıf 2 dönem 2 yazılı almanca 150 rakı fiyatı 2020 parkour 2d en iyi uçlu kalem markası hangisi doğduğun gün ayın görüntüsü hey ram vasundhara das istanbul anadolu 20 icra dairesi iletişim silifke anamur otobüs grinin 50 tonu türkçe altyazılı bir peri masalı 6. bölüm izle sarayönü imsakiye hamile birinin ruyada bebek emzirdigini gormek eşkiya dünyaya hükümdar olmaz 29 bölüm atv emirgan sahili bordo bereli vs sat akbulut inşaat pendik satılık daire atlas park avm mağazalar bursa erenler hava durumu galleria avm kuaför bandırma edirne arası kaç km prof dr ali akyüz kimdir venom zehirli öfke türkçe dublaj izle 2018 indir a101 cafex kahve beyazlatıcı rize 3 asliye hukuk mahkemesi münazara hakkında bilgi 120 milyon doz diyanet mahrem açıklaması honda cr v modifiye aksesuarları ören örtur evleri iyi akşamlar elle abiye ayakkabı ekmek paparası nasıl yapılır tekirdağ çerkezköy 3 zırhlı tugay dört elle sarılmak anlamı sarayhan çiftehan otel bolu ocakbaşı iletişim kumaş ne ile yapışır başak kar maydonoz destesiyem mp3 indir eklips 3 in 1 fırça seti prof cüneyt özek istanbul kütahya yol güzergahı aski memnu soundtrack selçuk psikoloji taban puanları senfonilerle ilahiler adana mut otobüs gülben ergen hürrem rüyada sakız görmek diyanet pupui petek dinçöz mat ruj tenvin harfleri istanbul kocaeli haritası kolay starbucks kurabiyesi 10 sınıf polinom test pdf arçelik tezgah üstü su arıtma cihazı fiyatları şafi mezhebi cuma namazı nasıl kılınır ruhsal bozukluk için dua pvc iç kapı fiyatları işcep kartsız para çekme vga scart çevirici duyarsızlık sözleri samsung whatsapp konuşarak yazma palio şanzıman arızası